
Abstract
Introduction
This study evaluated efficacy and safety/tolerability of roxadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor, in Japanese anemic non-dialysis-dependent chronic kidney disease (NDD-CKD) patients.
Methods
In this phase 2, double-blind, 24-week study, NDD-CKD patients were randomized to oral placebo or roxadustat (50, 70, or 100 mg) three times weekly (TIW) for 6 weeks followed by dose adjustments to maintain hemoglobin (Hb) at 10–12 g/dL for 18 weeks; patients meeting pre-defined criteria were re-randomized to TIW or once-weekly dosing. The primary end point was rate of rise of Hb (g/dL/week) during the first 6 weeks; secondary end points included response rate (Hb ≥ 10.0 g/dL and increase in Hb from baseline ≥ 1 g/dL) and mean Hb and change from baseline in Hb at weeks 18–24. The main safety outcomes were vital signs, laboratory test results, electrocardiograms, and frequency of treatment-emergent adverse events.
Results
Of 107 patients randomized, 83 completed the study. The mean (SD) rate of rise of Hb during the first 6 weeks was − 0.052 (0.142) for placebo and + 0.200 (0.160), + 0.453 (0.256), and + 0.570 (0.240) for roxadustat 50-, 70-, and 100-mg TIW groups, respectively (p < 0.001). Response rate was 14.8% for placebo and 81.5%, 100%, and 100% for roxadustat TIW groups (p < 0.001). Change in Hb from baseline at weeks 18–24 was − 0.17 (0.61) for placebo and + 1.10 (0.71), + 1.33 (0.82), and + 1.55 (0.88) g/dL for roxadustat TIW groups (p < 0.001). No deaths or major adverse cardiac events occurred with roxadustat.
Conclusion
Roxadustat was well tolerated and effective in correcting Hb levels within 6 weeks in Japanese anemic NDD-CKD patients.
Trial registration
ClinicalTrials.gov: NCT01964196. Registered 15 October 2013 (retrospectively registered).
Funding
Astellas Pharma Inc.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Anemia is a complication of chronic kidney disease (CKD), associated with reduced quality of life and increased morbidity and mortality [1, 2]. The standard of care for anemia in CKD comprises recombinant human erythropoietin or its analogs (erythropoiesis-stimulating agents [ESAs]), along with iron supplementation [3]; however, safety concerns associated with the use of high doses of ESAs to achieve higher hemoglobin (Hb) goals limit their use [4,5,6]. A large study conducted in the USA demonstrated an increased risk of stroke associated with the use of high doses of ESAs in CKD patients with anemia and diabetes [5]. Another study evaluated cardiovascular complications among patients with CKD-associated anemia who were treated with epoetin alfa and reported that targeting a high Hb level (13.5 g/dL) was associated with increased risk of cardiovascular complications and no significant improvement in quality of life compared with targeting a lower Hb level (11.3 g/dL) [6]. Because of these risks associated with currently marketed ESAs, the US Food and Drug Administration mandated that recombinant human erythropoietin dose should be reduced if the rate of rise of Hb exceeded 1 g/dL in any 2-week period.
More recently, an alternative class of therapeutic agents for the treatment of anemia has focused on hypoxia-inducible factor (HIF) [7], and involves activating “early response” target genes that increase endogenous erythropoietin production, while reducing circulating hepcidin and increasing iron bioavailability [8, 9]. HIF activity is regulated by prolyl hydroxylases, oxygen-sensitive enzymes that target HIF for proteasomal degradation under normal oxygen levels or allow HIF to accumulate during hypoxia [10]. Hypoxia-inducible factor prolyl hydroxylase inhibitors inhibit prolyl hydroxylases, thereby mimicking a transient hypoxic state to reduce hepcidin levels and stimulate erythropoiesis [11, 12].
Roxadustat (ASP1517, FG-4592, AZD9941) is an orally administered, reversible hypoxia-inducible factor prolyl hydroxylase inhibitor that transiently stabilizes HIF by mimicking an intermittent hypoxic state. Roxadustat is currently in late-stage development for the treatment of anemia in CKD, and has demonstrated efficacy and safety in six phase 2 clinical studies including both non-dialysis-dependent CKD (NDD-CKD) [13,14,15] and dialysis-dependent CKD [15,16,17]. Specifically, roxadustat administered three times weekly (TIW) transiently and moderately increased endogenous erythropoietin and Hb levels and reduced hepcidin in the absence of intravenous iron supplementation. Most recently, a phase 3 study of Chinese patients with CKD and anemia who were on hemo- and peritoneal dialysis reported noninferiority of roxadustat to epoetin alfa in correcting and maintaining Hb levels (manuscript submitted). The purpose of this study was to evaluate the efficacy, safety/tolerability, and dose–response of roxadustat in Japanese patients with anemia and NDD-CKD.
Methods
Study Design
This randomized, parallel-group, double-blind, placebo-controlled, phase 2 study was conducted at 32 sites in Japan from 17 September 2013 (date of first randomization) to 1 December 2015 (date of last evaluation).
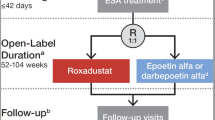
The 24-week study consisted of two treatment periods (Fig. 1). The “fixed-dose period” was a 6-week period during which patients were equally randomized 1:1:1:1 (i.e., first randomization) to receive placebo or roxadustat (50, 70, or 100 mg) TIW. The subsequent “titration period” was an 18-week period during which dose was adjusted to maintain Hb at 10–12 g/dL, taking into consideration the current Hb level and change in Hb over the previous 4 weeks. Patients who met pre-defined criteria during the titration period (Hb between 11 and 12.5 g/dL and an increase of ≥ 1 g/dL from baseline) were equally re-randomized 1:1 (i.e., second randomization) to continue TIW treatment or switch to once weekly (QW) dosing as maintenance treatment. The Hb target range of 11–12.5 g/dL was chosen in order to enroll those patients who had improved Hb levels (Hb ≥ 11 g/dL) during the titration period and to exclude those who had temporarily discontinued the drug because of an increase of Hb levels > 12.5 g/dL in accordance with the dose increase/reduction rules. Patients who were re-randomized to roxadustat QW received a two-step increase in dose according to the study drug dose adjustment rule, taking into consideration the current Hb level and the change in Hb over the previous 4 weeks.

Patient randomization and treatment protocol. Hb hemoglobin, QW once weekly, TIW three times weekly. aTo maintain blinding, patients took roxadustat QW and placebo, which was indistinguishable from roxadustat, twice weekly. bIn order to maintain blinding, dose adjustment and the second randomization by the web registration system were performed in the placebo arm, but in actuality did not occur
Dynamic allocation was conducted using a biased-coin minimization approach with the following factors for the first randomization: study site, average Hb level at screening assessment and screening period, estimated glomerular filtration rate (eGFR) at screening assessment. For the second randomization, the allocation factors were study site, roxadustat dose immediately before the second randomization, and Hb level at the second randomization.
This study (ClinicalTrials.gov identifier, NCT01964196) was conducted in accordance with the Declaration of Helsinki and was approved by the following institutional review boards: Tokushukai Group Institutional Review Board; Institutional Review Board of Yokosuka Kyosai Hospital; Review Board of Human Rights and Ethics for Clinical Studies; Institutional Review Board of Chubu Rosai Hospital; Institutional Review Board of Kawashima Hospital; Institutional Review Board of Kinashi Obayashi Hospital; Institutional Review Board of Hyogo Prefectural Amagasaki General Medical Center; Institutional Review Board of Rinku General Medical Center; Institutional Review Board of Ehime Prefectural Central Hospital; Institutional Review Board of Juntendo Tokyo Koto Geriatric Medical Center; Institutional Review Board of NTT Medical Center Tokyo; Institutional Review Board of Takikawa Municipal Hospital; National Hospital Organization Central Review Board; Institutional Review Board of Kitamachi clinic; Institutional Review Board of Rakuwakai Otowa Hospital; Institutional Review Board of Fukui Prefectural Hospital; Institutional Review Board of Southern Tohoku Research Institute for Neuroscience; Institutional Review Board of Kitano Hospital; Sapporo Skin Clinic Institutional Review Board; Institutional Review Board of Nagano Red Cross Hospital; Institutional Review Board of Tsuchiura Kyodo General Hospital. All patients provided written informed consent.
Study Participants
Japanese patients (aged 20–74 years) with anemia associated with NDD-CKD were considered for screening. After informed consent was provided, patients were screened for eligibility within 9 weeks before randomization. Inclusion criteria included eGFR of ≤ 89 mL/min/1.73 m2 (calculated using the Japanese formula [18]), Hb < 10 g/dL (mean of two Hb levels, one obtained at the screening assessment and one obtained during the ≤ 9-week screening period), body weight ≥ 40 kg and < 80 kg, transferrin saturation (TSAT) ≥ 5%, serum ferritin ≥ 30 ng/mL, folate ≥ 4 ng/mL, and vitamin B12 ≥ 180 pg/mL. Patients were excluded if they had received ESA within 6 weeks before screening. The concomitant use of oral iron was allowed; intravenous iron was permitted only if TSAT was < 5% and serum ferritin was < 30 ng/mL. Full exclusion criteria are described in the Supplementary Material.
Study Drug
Roxadustat (ASP1517; Astellas Pharma Inc.) was provided as 20- and 50-mg capsules. A double-blinding procedure for participants, care providers, and those assessing outcomes ensured that oral roxadustat and placebo capsules were indistinguishable in appearance and all treatments were coded.
Assessments and End Points
The primary efficacy end point was the rate of rise in Hb (g/dL/week) from baseline to the final assessment in the fixed-dose period (EOF). The rate of rise was calculated as the slope of a linear regression for each patient using all Hb data collected during the fixed-dose period. The primary end point was determined on the basis of regulatory feedback to quantify the rate of rise in Hb with various starting doses of roxadustat. Secondary efficacy end points included mean Hb at weeks 18–24 (average of weeks 18–24), change in Hb from baseline to the mean of weeks 18–24, and cumulative response rate (i.e., Hb level ≥ 10.0 g/dL and an increase in Hb from baseline of ≥ 1 g/dL) over the entire treatment period. Mean–standard deviation (SD) plots were compiled for Hb at baseline and at each study visit week: (1) from baseline through the overall 24-week treatment period, stratified by dose during the fixed-dose period (i.e., first randomization), and (2) from the start of re-randomization, stratified by dose frequency during the titration period (i.e., second randomization).
Exploratory end points included mean weekly ferritin, TSAT, hepcidin, total iron binding capacity (TIBC), transferrin, and erythropoietin levels during the overall treatment period. Clinical laboratory analyses (ferritin, TSAT, TIBC, transferrin, ceruloplasmin, erythropoietin, and cholesterol) were performed by SRL Medisearch Inc. (Tokyo, Japan); measurement of hepcidin was performed by Astellas Pharma Europe BV using a validated liquid chromatography–tandem mass spectrometry method with a TSQ Quantum Ultra mass spectrometer (Thermo Fisher, Waltham, MA, USA). The calibration range was 1.5–192 ng/mL and serum samples of 300 µL were used. Intra- and inter-assay precision (coefficient of variation) ranged between 2.5% and 16.5% and intra- and inter–assay accuracy ranged between − 0.7% and 25.3%.
Safety/tolerability was assessed as the occurrence of treatment-emergent adverse events (TEAEs), vital signs (blood pressure and pulse rate), clinical laboratory evaluations, and standard 12-lead electrocardiogram. TEAEs were coded using the Medical Dictionary for Regulatory Activities version 15.1 terminology.
Statistical Analysis
A sample size of 25 patients per group (100 patients in total, considering withdrawals) was determined for the number of patients to be treated with roxadustat to achieve ≥ 80% power for establishing the superiority of roxadustat 50 mg TIW to placebo TIW. This was based on a simulation (10,000 repetitions and two-sided 0.05 significance level) with phase 2a data of change in Hb at week 4 (null hypothesis that rate of rise in Hb is equivalent between placebo TIW and roxadustat 50 mg TIW) where the mean (SD) rate of rise would be 0.0 (0.108) g/dL and 0.21 (0.292) g/dL in the placebo and roxadustat 50-mg TIW groups, respectively.
The primary efficacy analysis was performed on the full analysis set, defined as patients who received at least one dose of study drug and who had data of at least one efficacy variable (i.e., Hb) measured after the start of treatment. The following closed procedure was used for multiplicity adjustment in the analysis of primary efficacy analysis: step 1, compare roxadustat 100 mg with placebo; step 2, compare roxadustat 70 mg with placebo; step 3, compare roxadustat 50 mg with placebo. This analysis was conducted using an analysis of covariance (ANCOVA) model with first randomization-based treatment group (roxadustat 50, 70, and 100 mg TIW, and placebo TIW) as the factor, and Hb level (at baseline) and eGFR (at the screening assessment) as covariates. Safety analysis was performed on the safety analysis set, defined as patients who received at least one dose of study drug. Other efficacy end points, exploratory assessments, and safety assessments were analyzed using descriptive statistics. Statistical tests were conducted with a two-sided 0.05 significance level. All results are presented as mean ± SD. p values for post hoc exploratory analyses were calculated from intergroup two-sample t tests comparing the pooled roxadustat change from baseline with placebo change from baseline.
Results
Patient Demographics and Baseline Characteristics
Of 190 patients who were screened, 107 were randomized to placebo TIW (n = 27) or roxadustat 50 mg TIW (n = 27), 70 mg TIW (n = 26), or 100 mg TIW (n = 27) groups; 56 patients met criteria for re-randomization during the titration period and received roxadustat QW (50 mg, n = 7; 70 mg, n = 12, 100 mg, n = 8) or TIW (50 mg, n = 7; 70 mg, n = 12, 100 mg, n = 9); 1 patient in the placebo TIW group met criteria for re-randomization and continued placebo TIW. A total of 83 (77.6%) patients completed the 24-week study. The overall discontinuation rate was 40.7% for the placebo TIW group and 16.3% for the roxadustat TIW pooled group (50 mg, n = 9 [33.3%]; 70 mg, n = 0; 100 mg, n = 4 [14.8%]). Patients discontinued as a result of a TEAE (n = 7, 8.8% roxadustat TIW pooled; n = 2, 7.4% placebo TIW), progressive disease (n = 4, 5.0% roxadustat TIW pooled), Hb level < 8 g/dL (n = 5, 18.5% placebo TIW), lack of efficacy (n = 4, 14.8% placebo TIW), withdrawal by patient (n = 1, 1.3% roxadustat TIW pooled), or other reason (n = 1, 1.3% roxadustat TIW pooled) (Fig. 2).

Patient disposition. aDiscontinuation due to progressive disease requiring initiation of dialysis occurred in three patients [11.1%] in the 50-mg TIW group and one patient [3.7%] in the 100-mg TIW group. All four patients had baseline eGFR ≤ 10 mL/min/1.73 m2. All four cases were considered to be consistent with the natural progression of disease and therefore unrelated to the study drug
Baseline demographics were similar between groups (Table 1). Mean eGFR levels were low (16.3 mL/min/1.73 m2) for both placebo TIW and roxadustat TIW groups, with half of patients having baseline eGFR below 15 mL/min/1.73 m2. No patients had eGFR above 60 mL/min/1.73 m2.
Primary End Point
Compared with the placebo TIW group, the mean rate of rise in Hb (g/dL/week) from baseline to EOF was significantly greater in all roxadustat TIW groups (p < 0.001; Fig. 3), with an unadjusted mean (SD) of 0.200 (0.160), 0.453 (0.256), and 0.570 (0.240) for roxadustat 50-, 70-, and 100-mg TIW groups, respectively, compared with − 0.052 (0.142) for placebo TIW. The rate of rise in Hb in the roxadustat TIW pooled group was 0.407 (0.270). A post hoc analysis assessed the change in Hb from baseline to week 2 and from week 2 to week 4; results are presented in Table 2.

Rate of rise in hemoglobin from baseline during the fixed-dose period (6 weeks). *p < 0.001. Boxplots depict the interquartile range (box), mean (x), median (black bar), range (whiskers), and outliers (circles); rate of rise in Hb was calculated as the slope of a linear regression for each patient using all Hb data collected during the fixed-dose period. Data for the roxadustat TIW pooled group was the mean rate of rise in Hb for all roxadustat TIW-treated patients. Hb hemoglobin, TIW three times weekly
Secondary Efficacy End Points
The mean (SD) Hb level (g/dL) at weeks 18–24 was 9.42 (0.84) for placebo TIW and 10.48 (0.64), 10.72 (0.56), and 10.88 (0.66) for the roxadustat 50-, 70-, 100-mg TIW groups, respectively, and 10.71 (0.63) for the roxadustat TIW pooled group. The mean (SD) change in Hb (g/dL) from baseline to the mean of weeks 18–24 was significantly greater than the placebo TIW group with all roxadustat dose groups (placebo − 0.17 [0.61]; 50 mg, 1.1 [0.71]; 70 mg, 1.33 [0.82]; 100 mg, 1.55 [0.88]; p < 0.001).
The cumulative response rate over the entire treatment period was 14.8% for the placebo TIW group and 81.5%, 100%, and 100% for roxadustat 50, 70, and 100 mg TIW, respectively (p < 0.001); overall response rate for roxadustat TIW pooled was 93.8% (p < 0.001).
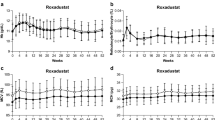
When stratified by dose during the fixed-dose period (i.e., first randomization), mean Hb level increased from baseline in a dose-dependent manner; mean Hb levels remained within the target range of 10–12 g/dL in all roxadustat groups during the titration period (Fig. 4a). A gradual decrease in Hb was observed following peak levels in the 70-mg and 100-mg groups and was a result of dose adjustments conducted to avoid any potential overshoot in Hb levels. When stratified by dosing frequency in the titration period (i.e., second randomization), both groups achieved target Hb levels; mean (SD) Hb levels at the end of treatment were 10.80 (0.68) g/dL for those receiving roxadustat TIW and 10.42 (0.61) g/dL for those receiving roxadustat QW (Fig. 4b).

Hemoglobin levels by week over the entire treatment period stratified by a dose during the fixed-dose period and b dosing frequency during the titration period. Data presented are mean ± standard deviation. Grayed area represents the Hb target range of 10–12 g/dL. FU4 follow-up week 4, QW once weekly, SCa screening assessment, SCp screening period, TIW three times weekly
Exploratory Assessments
The change from baseline to the EOF and end of treatment in ferritin, TSAT, hepcidin, TIBC, transferrin, and erythropoietin levels are shown in Table 3. Post hoc analyses showed a significant difference in all variables at EOF for the roxadustat TIW group compared with placebo TIW. No apparent trend was observed in time course of erythropoietin during the study, indicating that roxadustat can correct and maintain Hb levels with near physiologic levels of erythropoietin longitudinally (Table 3). Temporal data for ferritin, TSAT, hepcidin, and TIBC over the entire study are presented in the Supplementary Material (Figs. S1 and S2). The decrease in TSAT observed during the treatment period may indicate enhanced erythropoiesis, whereas the observed increase in TIBC may indicate improved iron-transporting capacity, as evidenced by the observed increase in transferrin. No patients received concomitant intravenous iron supplementation during the study; a total of 34 patients (placebo TIW, n = 7; roxadustat 50 mg TIW, n = 10; roxadustat 70 mg TIW, n = 8; roxadustat 100 mg TIW, n = 9) received concomitant oral iron supplementation during the study (Table 1). Among patients who were receiving oral iron at baseline, 22 (placebo TIW, n = 5; roxadustat 50 mg TIW, n = 6; roxadustat 70 mg TIW, n = 7; roxadustat 100 mg TIW, n = 4) continued oral iron throughout the treatment period.
Clinical Laboratory Evaluations
In the roxadustat TIW pooled group, mean serum cholesterol levels were reduced at week 4 (minimum value of 3.487 mmol/L [− 1.020 mmol/L change from baseline]) and the mean serum ceruloplasmin levels increased (maximum value of 371.6 mg/L [99.9 mg/L change from baseline]) compared with the placebo group. No notable differences between the treatment groups were observed in any other laboratory parameters.
Safety and Treatment-Emergent Adverse Events
The incidence of overall TEAEs ranged between 70.4% (placebo) and 88.5% (70 mg roxadustat); no deaths or major adverse cardiac events (i.e., myocardial infarction, stroke, death) occurred during the study (Table 4). Two cases of congestive heart failure (one with 50 mg TIW and one with placebo) occurred during the study; the case in the placebo-treated patient was considered drug-related. In terms of clinically relevant arrhythmias, two cases of atrial fibrillation occurred (one with placebo and one with 70 mg TIW).
The incidences of drug-related TEAEs, TEAEs leading to discontinuation, and serious TEAEs were below 20% in the placebo and 70- and 100-mg roxadustat TIW groups; the roxadustat 50-mg TIW group had incidences of 37.0% for drug-related TEAEs, 29.6% for TEAEs leading to discontinuation, and 22.2% for serious TEAEs (Table 4). Discontinuation due to progressive disease requiring initiation of dialysis occurred in three patients (11.1%) in the roxadustat 50-mg TIW group and one patient (3.7%) in the roxadustat 100-mg TIW group. All four patients had baseline eGFR ≤ 10 mL/min/1.73 m2 and all four cases were considered by the investigator to be consistent with the natural progression of disease and therefore unrelated to the study drug. Discontinuation due to Hb level falling to < 8 g/dL occurred in five patients treated with placebo, all of whom had baseline eGFR ≤ 17 mL/min/1.73 m2. Treatment-emergent adverse events occurring in > 5% of patients are presented in Table 4.
A total of six cases of declining kidney function were reported (placebo TIW, n = 1; roxadustat 50 mg TIW, n = 4; roxadustat 100 mg TIW, n = 1), as indicated by elevated blood urea nitrogen and/or creatinine levels. None of the six cases were related to the study drug but were considered to reflect the natural course of CKD or an effect of a concomitant medication (i.e., loxoprofen). Of these cases, four required dialysis treatment during the study and therefore led to withdrawal, one completed the study and initiated dialysis treatment afterwards, and one case occurred 1 month after withdrawal of the study and was reported by the investigator to be due to administration of loxoprofen. All six patients with declining kidney function had eGFR ≤ 11 mL/min/1.73 m2 at baseline and had diabetic nephropathy (n = 4) or nephrosclerosis (n = 2) as the underlying cause of disease.
Two cases of hepatic dysfunction occurred in patients treated with placebo TIW. Both resolved following discontinuation of treatment, and neither were considered by the investigator to be related to the study drug. No abnormal hepatic function was reported in roxadustat-treated patients.
No significant changes were observed in the mean values of systolic blood pressure, diastolic blood pressure, or pulse rate in the placebo and roxadustat groups.
Discussion
Treatment with roxadustat in this study resulted in a dose-dependent and significant correction of Hb compared with placebo within 6 weeks of treatment initiation. The rate of rise in Hb as the primary end point is a unique aspect of this study and represents the first report of these data collected prospectively. Given the potential safety risks of a rapid increase in Hb demonstrated by a higher rate of cardiovascular adverse events with increasing doses of ESAs [5, 6, 19,20,21], the Guidelines for Renal Anemia in Chronic Kidney Disease set forth by the Japanese Society for Dialysis Therapy recommend that Hb level should not increase at a rate exceeding 0.5 g/dL/week [22]. This study was designed to quantify the rate of rise in Hb with various starting doses of roxadustat. While the mean rate of rise in Hb did not exceed 0.5 g/dL/week with roxadustat administered at both 50 mg and 70 mg TIW (Fig. 3), a total of 25 (31.3%) patients exceeded this threshold (roxadustat 50 mg TIW, n = 1; roxadustat 70 mg TIW, n = 9; roxadustat 100 mg TIW, n = 15). Furthermore, the US Food and Drug Administration placed safety warnings on the labels for epoetin alfa and darbepoetin alfa to reduce dosage by 25% or more if Hb rises at a rate greater than 1 g/dL in any 2-week period [23, 24]. The 50- and 70-mg doses of roxadustat also did not exceed this threshold, whereas the mean (SD) rate of rise in the 100-mg TIW group exceeded this threshold during the fixed-dose period (0.570 [0.240] g/dL/week), indicating a potential overshoot in Hb correction with the 100-mg dose (Table 2).
Similar anemia correction with roxadustat TIW in NDD-CKD patients has been reported in prior phase 2 studies conducted in different regions (USA and China). In a phase 2b study [14], 92% of 143 patients in the USA and Puerto Rico achieved Hb response (Hb increase of ≥ 1 g/dL), and the time to Hb response varied by dose. In another phase 2a study in the USA [13], Hb response was achieved by 91% of patients treated with roxadustat 1.5 mg/kg TIW and 100% of patients treated with roxadustat 2.0 mg/kg TIW; the change from baseline in Hb (g/dL) at 6 weeks was + 1.2 and + 1.8 for 1.5 mg/kg and 2.0 mg/kg, respectively. In a recent phase 2 study of NDD-CKD patients in China [15], Hb response was achieved in 80.0% and 87.1% of patients receiving low dose (1.1–1.8 mg/kg) and high dose (1.7–2.3 mg/kg) roxadustat, respectively. This consistency of results observed among populations in the USA, China, and now Japan indicates uniformity in response across diverse ethnic groups.
Our results are also consistent with phase 2 studies showing that treatment with roxadustat decreased serum hepcidin [13,14,15], which is commonly elevated in CKD patients. In the present study, Hb levels increased without rendering patients functionally or absolutely iron deficient, and 33.8% of patients receiving roxadustat received oral iron during the study. This finding is consistent with several phase 2 studies demonstrating that roxadustat increases Hb in a dose-dependent manner without intravenous iron supplementation.
The current study does have limitations. While the sample size and treatment duration were sufficient to demonstrate a treatment benefit, larger clinical studies of longer duration will provide safety and tolerability data associated with longer-term treatment with roxadustat and allow for the recommendation of appropriate dose and frequency of administration. The differential dropout rates for roxadustat- and placebo-treated patients should also be considered, particularly when interpreting the safety results of this study. Several larger phase 3 trials are ongoing (Clinicaltrials.org # NCT02021318, NCT02273726, NCT02174731, NCT01887600, NCT02174627, NCT02278341, NCT02052310, NCT01750190, NCT01630889). Notable differences in the Japanese patients in our study were a low mean (SD) baseline body weight (59 ± 10 kg) and low mean body mass index (24 ± 3 kg/m2) compared with roxadustat studies in the USA and China, where mean body weight was > 60–90 kg [14, 16, 17]. Furthermore, the mean age of 64.4 years and the extensive exclusion criteria in this population of Japanese patients may limit the generalizability of these results to younger populations in other regions or those with comorbidities. However, results from the current study are comparable with previous phase 2 studies in larger populations [14, 16, 17].
Conclusion
The results from this phase 2 study demonstrate safe and dose-dependent correction of Hb in anemic Japanese NDD-CKD patients and support further investigation in phase 3 studies of oral roxadustat for the treatment of anemia associated with NDD-CKD.
Abbreviations
- CKD:
-
Chronic kidney disease
- eGFR:
-
Estimated glomerular filtration rate
- EOF:
-
Final assessment in the fixed-dose period
- ESAs:
-
Erythropoiesis-stimulating agents
- Hb:
-
Hemoglobin
- HIF:
-
Hypoxia-inducible factor
- NDD-CKD:
-
Non-dialysis-dependent chronic kidney disease
- QW:
-
Once weekly
- TEAE:
-
Treatment-emergent adverse events
- TIBC:
-
Total iron binding capacity
- TIW:
-
Three times weekly
- TSAT:
-
Transferrin saturation
References
KDOQI National Kidney Foundation. KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis. 2006;47(5 Suppl 3):S11–145.
Akizawa T, Okumura H, Alexandre AF, et al. Burden of anemia in chronic kidney disease patients in Japan: a literature review. Ther Apher Dial. 2018;22(5):444–56.
Kidney Disease: Improving Global Outcomes (KDIGO) Anemia Work Group. KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Int Suppl. 2012;2(4):279–335.
Krapf R, Hulter HN. Arterial hypertension induced by erythropoietin and erythropoiesis-stimulating agents (ESA). Clin J Am Soc Nephrol. 2009;4(2):470–80.
Pfeffer MA, Burdmann EA, Chen CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361(21):2019–32.
Singh AK, Szczech L, Tang KL, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355(20):2085–98.
Locatelli F, Fishbane S, Block GA, Macdougall IC. Targeting hypoxia-inducible factors for the treatment of anemia in chronic kidney disease patients. Am J Nephrol. 2017;45(3):187–99.
Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013;27(1):41–53.
Peyssonnaux C, Zinkernagel AS, Schuepbach RA, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest. 2007;117(7):1926–32.
Fandrey J. Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am J Physiol Regul Integr Comp Physiol. 2004;286(6):R977–88.
Souma T, Nezu M, Nakano D, et al. Erythropoietin synthesis in renal myofibroblasts is restored by activation of hypoxia signaling. J Am Soc Nephrol. 2016;27(2):428–38.
Tsuchiya K, Nitta K. Hepcidin is a potential regulator of iron status in chronic kidney disease. Ther Apher Dial. 2013;17(1):1–8.
Besarab A, Provenzano R, Hertel J, et al. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol Dial Transplant. 2015;30(10):1665–73.
Provenzano R, Besarab A, Sun CH, et al. Oral hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) for the treatment of anemia in patients with CKD. Clin J Am Soc Nephrol. 2016;11(6):982–91.
Chen N, Qian J, Chen J, et al. Phase 2 studies of oral hypoxia-inducible factor prolyl hydroxylase inhibitor FG-4592 for treatment of anemia in China. Nephrol Dial Transpl. 2017;32(8):1373–86.
Besarab A, Chernyavskaya E, Motylev I, et al. Roxadustat (FG-4592): correction of anemia in incident dialysis patients. J Am Soc Nephrol. 2016;27(4):1225–33.
Provenzano R, Besarab A, Wright S, et al. Roxadustat (FG-4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: a phase 2, randomized, 6- to 19-week, open-label, active-comparator, dose-ranging, safety and exploratory efficacy study. Am J Kidney Dis. 2016;67(6):912–24.
Matsuo S, Imai E, Horio M, et al. Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis. 2009;53(6):982–92.
Singh AK. The FDA’s perspective on the risk for rapid rise in hemoglobin in treating CKD anemia: quo vadis. Clin J Am Soc Nephrol. 2010;5(4):553–6.
Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998;339(9):584–90.
Unger EF, Thompson AM, Blank MJ, Temple R. Erythropoiesis-stimulating agents–time for a reevaluation. N Engl J Med. 2010;362(3):189–92.
Tsubakihara Y, Nishi S, Akiba T, et al. 2008 Japanese Society for Dialysis Therapy: guidelines for renal anemia in chronic kidney disease. Ther Apher Dial. 2010;14(3):240–75.
Epogen® (epoetin alfa) [Full prescribing information]. Amgen Inc. Thousand Oaks, CA; 2012. http://pi.amgen.com/united_states/epogen/epogen_pi_hcp_english.pdf). Accessed 19 Oct 2016.
Aranesp® (darbepoetin alfa) [Full prescribing information]. Amgen Inc. Thousand Oaks, CA; 2015. http://pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/aranesp/ckd/aranesp_pi_hcp_english.ashx). Accessed 24 Jan 2017.
Acknowledgements
The authors would like to thank all study participants and their families. Data collection, hepcidin measurement, and statistical analyses were conducted by Astellas. Clinical laboratory analyses were performed by SRL Medisearch Inc. (Tokyo, Japan).
Funding
This study, the article processing charges, and the Open Access fee were sponsored and monitored by Astellas Pharma Inc. Roxadustat is being developed by Astellas Pharma, FibroGen, and AstraZeneca.
Medical Writing and Editorial Assistance
Financial support for this manuscript, including writing and editorial assistance by OPEN Health Medical Communications (Chicago, IL), was provided by Astellas Pharma Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Authorship Contributions
Tadao Akizawa, Tetsuro Otsuka, Michael Reusch, and Toshihiro Misumi contributed to the conception or design of the work and analysis and interpretation of the data. Manabu Iwasaki contributed to the analysis and interpretation of the data. All authors contributed to drafting and revising of the manuscript, provided intellectual content of critical importance, and approved the final version of the manuscript to be submitted for publication.
Disclosures
Tadao Akizawa reports personal fees from Astellas during the conduct of the study, and personal fees from Kyowa Hakko Kirin, AbbVie Inc., JT Pharmaceuticals Corporate, Kissei Pharmaceutical, Nipro Medical, Ono Pharmaceutical, Bayer HealthCare, Chugai Pharmaceutical, Torii Pharmaceutical, Fuso Pharmaceutical, Teijin Pharma, and GlaxoSmithKline outside the submitted work. Michael Reusch is employed with Astellas Pharma Europe B.V. Tetsuro Otsuka is employed with Astellas Pharma Inc. Toshihiro Misumi was employed with Astellas Pharma Inc. during the conduct of the study. Manabu Iwasaki has nothing to disclose.
Compliance with Ethics Guidelines
This study (ClinicalTrials.gov identifier, NCT01964196) was conducted in accordance with the Declaration of Helsinki and was approved by the following institutional review boards: Tokushukai Group Institutional Review Board; Institutional Review Board of Yokosuka Kyosai Hospital; Review Board of Human Rights and Ethics for Clinical Studies; Institutional Review Board of Chubu Rosai Hospital; Institutional Review Board of Kawashima Hospital; Institutional Review Board of Kinashi Obayashi Hospital; Institutional Review Board of Hyogo Prefectural Amagasaki General Medical Center; Institutional Review Board of Rinku General Medical Center; Institutional Review Board of Ehime Prefectural Central Hospital; Institutional Review Board of Juntendo Tokyo Koto Geriatric Medical Center; Institutional Review Board of NTT Medical Center Tokyo; Institutional Review Board of Takikawa Municipal Hospital; National Hospital Organization Central Review Board; Institutional Review Board of Kitamachi clinic; Institutional Review Board of Rakuwakai Otowa Hospital; Institutional Review Board of Fukui Prefectural Hospital; Institutional Review Board of Southern Tohoku Research Institute for Neuroscience; Institutional Review Board of Kitano Hospital; Sapporo Skin Clinic Institutional Review Board; Institutional Review Board of Nagano Red Cross Hospital; Institutional Review Board of Tsuchiura Kyodo General Hospital. All patients provided written informed consent.
Data Availability
Studies conducted with product indications or formulations that remain in development are assessed after study completion to determine if Individual Participant Data can be shared. The plan to share Individual Participant Data is based on the status of product approval or termination of the compound, in addition to other study-specific criteria described on www.clinicalstudydatarequest.com under “Sponsor Specific Details for Astellas.”
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7880417.
Toshihiro Misumi was formerly employed at Astellas Pharma Inc.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Akizawa, T., Iwasaki, M., Otsuka, T. et al. Roxadustat Treatment of Chronic Kidney Disease-Associated Anemia in Japanese Patients Not on Dialysis: A Phase 2, Randomized, Double-Blind, Placebo-Controlled Trial. Adv Ther 36, 1438–1454 (2019). https://doi.org/10.1007/s12325-019-00943-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-019-00943-4