
Abstract
Introduction
Different prolonged-release formulations of tacrolimus are available. To date, the pharmacokinetic (PK) profile of LCP-tacrolimus (LCPT; Envarsus®) has not been compared with PR-Tac (Advagraf®) in de novo kidney transplant recipients. These profiles will guide clinical recommendations for the initiation and dose titration strategies of once-daily tacrolimus formulations.
Methods
This randomized, parallel-group, open-label, multicenter PK study randomized 75 de novo, adult, white kidney transplant recipients to LCPT 0.17 mg/kg/day (n = 37) or PR-Tac 0.20 mg/kg/day (n = 38) for 4 weeks. Dose adjustments were permitted to target a pre-defined therapeutic range based on measured trough blood concentrations.
Results
PK analysis (days 1, 3, 7 and 14) included 68 patients (LCPT, n = 33; PR-Tac, n = 35). Similar proportions of patients were within the pre-defined therapeutic tacrolimus trough blood concentration range, with < 12% in each group having below-target trough levels over the study period. LCPT demonstrated ~ 30% greater relative bioavailability [LCPT/PR-Tac adjusted geometric mean ratio: day 3, 1.32 (p = 0.007); day 7, 1.25 (p = 0.051); day 14, 1.43 (p = 0.002)] and ~ 30% lower peak-to-trough percentage fluctuation of blood concentration [LCPT/PR-Tac adjusted geometric mean ratio: day 3, 0.70 (p < 0.001); day 7, 0.68 (p < 0.001); day 14, 0.73 (p = 0.004)] in addition to longer time to maximum blood concentration (tmax), lower maximum concentration (Cmax) and a consistently lower daily dose (~ 40% dose reduction with LCPT vs. PR-Tac by day 28). Safety profiles were similar.
Conclusion
In de novo kidney transplant recipients, prolonged-release formulations of tacrolimus can reach therapeutic concentrations in the immediate post-transplant period. LCPT has greater relative bioavailability and lower peak-to-trough fluctuation compared with PR-Tac.
Trial registration
Registered at ClinicalTrials.gov; study number NCT02500212.
Funding
Chiesi Farmaceutici S.p.A.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tacrolimus is a potent calcineurin inhibitor used in almost all kidney transplant recipients [1] because of its proven ability to prevent graft rejection. However, the use of tacrolimus is complicated by a narrow therapeutic index that requires regular monitoring of the drug concentration in the blood to maintain therapeutic targets and minimize toxicity [2, 3]. Both under- and over-dosing of tacrolimus may compromise clinical outcomes by exposing patients to either the risks of graft rejection or adverse events (AEs) associated with immunosuppressive therapy [4]. Although tacrolimus trough levels are currently the most widely used pharmacokinetic (PK) parameter in the standard management of transplanted patients, a recent retrospective study on 488 renal transplant recipients showed several patient-specific factors, such as recipient’s age, BMI, hemoglobin level and the presence of anti-HCV antibodies, increase the sensitivity to tacrolimus, representing independent risk factors for potential tacrolimus-related toxicity. The authors highlight the importance of further studies to optimize management of risk-loaded patients [5].
Tacrolimus is available in a twice-daily immediate-release (IR-Tac; originator Prograf®, Astellas Pharma Inc., Tokyo, Japan) and recently in two once-daily prolonged-release formulations. PR-Tac (Advagraf®, Astellas Pharma Inc., Tokyo, Japan), the first prolonged-release formulation, showed similar efficacy to IR-Tac [6]; however, in the phase III study in de-novo renal transplantation, the rate of acute rejection was numerically higher with PR-Tac, and PR-Tac failed to demonstrate non-inferiority to IR-Tac at 24 weeks (treatment difference 4.5%, 95% CI − 1.8%, 10.9% with the upper limit outside the predefined 10% non-inferiority margin) [7].
LCP-tacrolimus (LCPT; Envarsus®, Chiesi Farmaceutici S.p.A., Parma, Italy) is a new prolonged-release formulation of tacrolimus, indicated for prophylaxis against transplant rejection in adult kidney or liver allograft recipients and for the treatment of allograft rejection in adult patients who are resistant to other immunosuppressants [8]. LCPT has been developed using MeltDose™ drug-delivery technology, which enhances oral bioavailability, controls drug release and produces a more distal distribution of tacrolimus within the gut [9, 10]. These features enable once-daily administration [11,12,13], and data from Phase I studies in healthy volunteers show that LCPT increases oral bioavailability and has lower peak-to-trough fluctuation compared with IR-Tac and PR-Tac, demonstrating that the two prolonged-release formulations are not bioequivalent [14, 15].
Multiple factors are known to affect the metabolism of tacrolimus, such as patient age, gender and gene polymorphisms in proteins involved in drug absorption and metabolism, such as CYP3A5 [16, 17]. In the ASERTAA study, conducted in African American kidney transplant recipients, the achievement of therapeutic trough concentrations with IR-Tac was associated with a 33% higher peak concentration in patients expressing the CYP3A5*1 allele compared with non-expressers (3*/3*) [18]. With LCPT, this difference was attenuated to 11%. The authors speculated that the minor susceptibility of LCPT to the CYP3A5 genotype may be because LCPT is absorbed in the more distal gastrointestinal tract where CYP3A5 activity is decreased [18]. LCPT also demonstrated non-inferiority in clinical outcomes and similar safety profiles to twice-daily tacrolimus in both de novo and stable kidney transplant patients [11,12,13].
Tremblay et al. published the first steady-state, head-to-head study (ASTCOFF) comparing the PK profiles of IR-Tac, PR-Tac and LCPT in stable kidney transplant recipients converting from established twice-daily tacrolimus therapy [9]. The study reported greater relative bioavailability, lower fluctuation and at least 30% dose reduction with LCPT versus IR-Tac and PR-Tac [9]. While ASTCOFF provided valuable insights into dose conversion strategies in stable patients, the PK profile of LCPT has not yet been compared with PR-Tac in de novo kidney transplant recipients. In addition, the PK of PR-Tac in de novo kidney transplant recipients is not currently well characterized, with discordant results among different studies [19]. The present study compared the PK profiles of LCPT and PR-Tac to explore whether a similar relationship also held in de novo patients and to guide clinical recommendations for the initiation and dose titration strategies of tacrolimus for de novo kidney transplant recipients.
Methods
The objectives of this study were to compare the PK and safety profile of once-daily LCPT or PR-Tac in adult de-novo kidney transplant recipients followed for 28 days after transplantation.
Study Population
Adult (≥ 18 years), white, de novo recipients of a living or deceased donor kidney transplant, including patients undergoing a second kidney transplant, with a body mass index (BMI) between 15 and 35 kg/m2, and who provided written informed consent were included in this analysis. Recipients of other transplants and patients who were already on immunosuppressants the day before transplantation were excluded.
Study Design and Sample Collection
This randomized, parallel-group, open-label, multicenter study (NCT02500212) was conducted at seven transplantation centers in France. Eligible patients were randomized 1:1 to either LCPT [Envarsus® 0.75 mg, 1 mg and 4 mg tablets (Veloxis Pharmaceuticals, Copenhagen, Denmark)] or PR-Tac [Advagraf® 0.5 mg, 1 mg, 3 mg, 5 mg capsules (Astellas Ireland Co., Ltd)] once-daily in the morning, initiated within 24 h after surgery, at starting doses of 0.17 mg/kg/day for LCPT and 0.20 mg/kg/day for PR-Tac. Starting doses were according to the respective EU summary of product characteristics [8, 20] and were maintained until day 3; thereafter, doses of LCPT and PR-Tac could be adjusted on the scheduled days (days 4, 8, 15 and 22) according to the trough levels measured on the previous day. Adjustments were performed with the aim of targeting predefined ranges of 5–15 ng/ml from days 2 to 15 and 5–10 ng/ml from days 16 to 28 (Fig. 1). A single whole-blood sample was collected at screening for CYP3A5 genotyping to identify single nucleotide polymorphisms (CYP3A5 *1/*1, *1/*3 and *3/*3). To avoid information on the patient genotype influencing dose adjustments, these data were disclosed at the end of the study. To study the PK profiles of tacrolimus in whole blood, 13 blood samples were collected in K2EDTA tubes pre-dose and 0.5, 1.0, 1.5, 2.0, 3.0, 4.0, 6.0, 8.0, 12.0, 16.0, 20.0 and 24.0 h after the morning dose on days 1, 3, 7 and 14. Tacrolimus trough levels were assessed in whole blood every day from day 2 to 8 and then on days 14, 15, 21 and 28 (Fig. 1). More detailed methods for genotyping and the assessment of tacrolimus whole-blood trough levels are provided in the Online Resource.

Study design
Compliance to tacrolimus prescription was evaluated based on the counts of tablets/capsules returned by the patients to each study visit and the patient’s diary used for recording drug intake starting from hospital discharge until the end of the study.
Concomitant immunosuppressive drugs were standardized to basiliximab (Simulect®, Novartis Pharmaceutical Corp., NJ, USA; 20 mg intravenously on day 0 and 4), mycophenolate mofetil (CellCept®, Hoffmann-La Roche Ltd., Mississauga, Ontario, Canada; 1 g orally twice daily until day 14 ± 2 days; 0.5 g orally twice daily until day 28 ± 2 days) and corticosteroids (intravenous methylprednisone: 10 mg/kg on day 0, 1 mg/kg on days 1 and 2; oral prednisone: 0.5 mg/kg on days 3–7 ± 2 days and then 0.3 mg/kg until day 15 ± 2 days and 10 mg until 28 ± 2 days).
A list of medications known to significantly interfere with the metabolism of tacrolimus was provided with the study protocol with the recommendation to avoid such medications unless clinically necessary.
PK Outcomes
The PK population was defined as all randomized subjects in the safety population (who had received ≥ one dose of study treatment) excluding subjects without any valid PK measurement. Patients with major protocol deviations significantly affecting PK were excluded from PK analysis for the period of the deviation, but not from the PK population.
All PK variables were calculated using non-compartmental analysis (NCA) with Phoenix WinNonlin 7.0 (Certara, Inc., Princeton, NJ, USA). The PK outcomes of this study were the ratio between the area under the curve from 0 to 24 h and daily dose (AUC0–24h)/daily dose) as an index of relative oral bioavailability, percentage peak-to-trough fluctuation [maximum whole-blood drug concentration (Cmax) − minimum whole-blood drug concentration (Cmin) × 100/average whole-blood drug concentration (Cavg)] and time to peak concentration (tmax) on days 1, 3, 7 and 14. Additional outcomes included AUC0– 24h, Cmax, Cavg and Cmin on days 1, 3, 7 and 14; trough levels [whole-blood trough drug concentration 24 h post dose (C24h)] daily from day 2 to day 8 and on days 14, 15, 21 and 28; proportion of patients with trough levels lower than, within and higher than the target range; proportion of patients with trough levels within the target range between day 2 to 4; and number of dose adjustments.
Safety Assessments
The safety population was defined as all randomized subjects who had received ≥ one dose of study treatment.
Safety parameters included vital signs, weight, BMI, serum creatinine, estimated glomerular filtration rate (eGFR; calculated according to the chronic Kidney Disease Epidemiology Collaboration creatinine equation [21]), blood chemistry, hematology, urinalysis, incidence of delayed graft function (DGF; defined as hemodialysis within 1 week following transplant or serum creatinine > 5 mg/dl by day 7), AEs, serious adverse events, adverse drug reactions (ADRs), serious ADRs, severe AEs, AEs leading to study discontinuation and AEs leading to death. Safety laboratory analyses were conducted at each site.
Statistical Analysis
No formal sample size calculations were undertaken. A sample size of 32 evaluable patients per treatment group was considered sufficient to characterize the PK of the study drugs. Based on a dropout rate of ~ 12%, 72 participants were planned (36 per treatment group).
All statistical analyses were performed using SAS 9.3 (SAS Institute Inc., Cary, NC, USA). Tacrolimus AUC0–24, C24h, Cmax, Cavg, Cmin, tmax, percentage peak-to-trough fluctuation [(Cmax − Cmin) × 100/Cavg] and AUC0–24h/daily dose were summarized using descriptive statistics by treatment and visit. Fluctuation (%) and AUC0–24h/daily dose at day 3, day 7 and day 14 were log-transformed to the natural logarithmic scale and then analyzed using an analysis of variance (ANOVA) model with treatment as a fixed effect. Alpha was set at 0.05, but the ratios of adjusted geometric means (GM) between the two groups were provided with 90% CIs. Due to the exploratory nature of the study, comparisons were done independently at three different time points without adjustment for any covariates and without accounting for repeated measures and multiple comparisons.
Tmax at day 3, day 7 and day 14 was analyzed using the Wilcoxon signed-rank test based on untransformed data. Hodges-Lehman estimates with 90% two-sided CIs for the median difference between the two treatments were applied.
The proportion of patients with tacrolimus C24h within the target range at day 2 and maintained within this range at day 3 and 4 was calculated by treatment. Differences between treatments were evaluated using Fisher’s exact test at the 0.05 significance level, with 95% CIs.
Compliance with Ethical Guidelines
All procedures performed in studies involving human participants were according to the clinical study protocol, the current International Council for Harmonization Good Clinical Practice guidelines, all local guidelines and the Declaration of Helsinki (1964 and amendments). The independent ethics committee/institutional research board for this study was the Committee for the Protection of Persons South Mediterranean V, CHU de Nice-Hôpital De Cimiez.
Informed consent was obtained from all individual participants included in the study. A specific Patient Information Sheet-Informed Consent Form for genotyping tests was obtained.
Results
Patient Disposition
A total of 76 patients were enrolled in this study between 30 June 2015 and 24 June 2016. Of the 75 patients randomized (98.7%), 37 and 38 patients were allocated to the LCPT and PR-Tac group, respectively. Six patients who did not receive any study drug (four in the LCPT group and two in the PR-Tac group) were excluded; one patient in the PR-Tac group was excluded from the PK population because of receiving treatment for only 1 day following diagnosis of vascular graft thrombosis on day 1. Therefore, 68 patients were evaluable for the PK analysis (LCPT group, n = 33; PR-Tac group, n = 35). Overall, 84.0% (n = 63) completed the study; of these, 83.8% (n = 31) and 84.2% (n = 32) were in the LCPT and PR-Tac group, respectively (Fig. 2).

Patient disposition. *Reasons for not receiving the allocated LCPT treatment (and being excluded from the safety and PK analysis populations) were: transplantation not performed (n = 2); patient did not meet inclusion/exclusion criteria (n = 1); study logistical issues (n = 1). Reasons for not receiving the allocated PR-Tac treatment (and being excluded from the safety and PK analysis populations) were: transplantation not performed (n = 1); patient withdrew consent (n = 1). ‡Death due to coronary artery thrombosis. †Discontinuation due to vascular graft thrombosis. §Excluded from the PK analysis for receiving only one dose of PR-Tac following the diagnosis of vascular graft thrombosis on study day 1. AE adverse event, LCPT LCP-tacrolimus, PK pharmacokinetic, PR prolonged release, CI confidence interval. Solid line represents 100%
One patient in each group received medication significantly interfering with the metabolism of tacrolimus: LCPT, fluconazole from day 12; PR-Tac, voriconazole from day 17. Both patients were excluded from PK analysis from the 1st day of the intake of the interfering concomitant medication. No patient was excluded from the study because of inadequate compliance.
Demographic characteristics varied slightly between the treatment groups, with a mean age of 58.9 and 53.7 years in the LCPT and PR-Tac group, respectively. The majority of patients (73.5%) were male in both treatment groups [LCPT, 66.7% (n = 22); PR-Tac, 80.6% (n = 28)]. All patients were white, and among those who provided consent for genotyping, 84.8% (n = 56) expressed CYP3A5 *3/*3, 15.2% expressed CYP3A5 *1/*3, and none expressed CYP3A5 *1/*1 (Table 1).
PK Outcomes
The relative bioavailability assessed as AUC0–24h/daily dose was greater (average of 33%) in the LCPT group than in the PR-Tac group on days 3, 7 and 14.
On day 1, the mean AUC0–24h/daily dose was similar between the treatment groups (19.02 ng h/ml/mg vs. 20.09 ng h/ml/mg for LCPT and PR-Tac, respectively). By day 3, the AUC0–24h/daily dose for the LCPT group had increased to 45.63 ng h/ml/mg compared with 34.60 ng h/ml/mg in the PR-Tac group, with a significant difference between groups (LCPT/PR-Tac 90% CI adjusted GM ratio 1.12–1.56, p = 0.007), which was maintained at day 7 and 14 (Table 2; Fig. 3).

Between treatment group PK comparisons: a analysis of % peak-to-trough fluctuation (PK population). b Analysis of dose-normalized AUC0–24h (PK population)
AUC0–24h/daily dose in patients with the CYP3A5 1*/3* genotype was numerically lower than that in CYP3A5 3*/3* genotype. However, the greater relative bioavailability in the LCPT group compared with the PR-Tac group was confirmed on each genotype subset on days 3, 7 and 14 (Online Resource).
Mean peak concentrations were consistently lower for LCPT compared with PR-Tac, leading to approximately 30% lower peak-to-trough percentage fluctuation compared with PR-Tac [LCPT/PR-Tac adjusted GM ratio: day 3, 70.17% (p < 0.001); day 7, 68.34% (p < 0.001); day 14, 72.6% (p = 0.004)] (Fig. 3). The lower peak-to-trough percentage fluctuation for LCPT compared with PR-Tac was maintained within each genotype subset (Online Resource).
LCPT had a consistently longer time to tmax compared with PR-Tac on days 1, 3, 7 and 14 [day 3 median (range) tmax 6.05 (1.5–24.0) vs. 2.00 (0.9–6.0) for LCPT vs. PR-Tac, respectively; Fig. 4). On all assessment days, LCPT also demonstrated a lower Cmax for tacrolimus, resulting in flatter PK profiles compared with the PR-Tac treatment group. At day 14, the Cmax ratio (90% CI) for LCPT compared with PR-TAC was 85.74 (74.36–98.85). Comparing LCPT with PR-Tac, the Hodges-Lehmann estimates of treatment effect for the time to tmax were 4.5 h (90% CI 4.0–5.8) on day 3, 3.6 h (90% CI 2.6–4.5) on day 7 and 3.0 h (90% CI 2.1–4.0) on day 14.

Concentration-time plots: a day 1; b day 3; c day 7; d day 14
Mean trough concentrations were generally similar between the treatment groups over the 28-day study period. The proportions of patients within the tacrolimus trough blood concentration target range over the study period were similar between the treatment groups over the 28-day study period (Online Resource Figure S1 and Table S1). A limited number of patients had below-target tacrolimus trough levels (< 12% in each group) between days 2 and 28. Overall, a greater proportion of CYP3A5 (1*/3*) expressers had tacrolimus trough blood concentrations below the target trough levels compared with non-expressers (3*/3*), particularly at day 2 where 50% of CYP3A5 expressers had tacrolimus trough blood concentrations below the target trough levels compared with 1.9% of non-expressers. At day 2, 33% of CYP3A5 expressers treated with LCPT had tacrolimus trough blood concentrations below the target trough levels compared with 75% receiving PR-Tac (Online Resource Table S1).
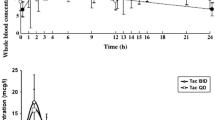
LCPT showed a consistently lower median daily dose with an approximately 50% lower dose by day 28 compared with the PR-Tac group [median daily dose (IQR) 5.00 mg (5.25) vs. 10.50 mg (9.00) for LCPT vs. PR-Tac, respectively; Fig. 5]. The number of dose adjustments was similar between the treatment groups over the study period: 74 adjustments (mean 2.24 per patient) in the LCPT group and 79 adjustments (mean 2.26 per patient) in the PR-Tac group.

Daily dose of tacrolimus: a mean daily dose; b weight-adjusted mean daily dose
Safety
Renal function improved over the 28-day treatment period in both groups. Mean (SD) serum creatinine levels decreased from 7.52 (3.02) mg/dl and 7.00 (2.40) mg/dl at baseline to 1.68 (0.41) mg/dl and 1.81 (0.69) mg/dl at day 28 for the LCPT and PR-Tac groups, respectively; mean change (SD) from baseline was − 5.90 (3.24) mg/dl for LCPT and − 5.31 (2.51) mg/dl for PR-Tac (Fig. 6a). Similar improvements were observed for eGFR, with increases observed from 7.68 (2.85) ml/min/1.73 m2 and 8.85 (3.86) ml/min/1.73 m2 at baseline to 43.73 (15.78) ml/min/1.73 m2 and 47.11 (22.03) ml/min/1.73 m2 at day 28 for LCPT and PR-Tac, respectively. Mean change in eGFR from baseline to day 28 was 36.62 (17.59) ml/min/1.73 m2 for LCPT and 37.47 (21.49) ml/min/1.73 m2 for PR-Tac (Fig. 6b).

Mean serum creatinine (a) and mean eGFR (b)
The proportion of patients with DGF was similar between the two groups, 39.5% (n = 13) in the LCPT group and 44.4% (n = 16) in the PR-Tac group [− 5.1% (95% CI − 28.6% to 18.5%); p = 0.80].
Treatment-emergent adverse events (TEAEs) are summarized in Table 3. A total of 274 TEAEs were reported in 33 patients in the LCPT group; 314 TEAEs were reported in 36 patients in the PR-Tac group. The most frequently reported TEAEs in both the LCPT and PR-Tac groups were anemia (57.6% and 55.6%, respectively), constipation (30.3% and 38.9%, respectively) and hematuria (30.3% and 38.9%, respectively). Serious TEAEs were reported in 15 (45.4%) and 11 (30.6%) patients in the LCPT and PR-Tac groups, respectively. ADRs were reported in ten (30.3%) and nine patients (25%) patients in the LCPT and PR-Tac group, respectively. The safety profile of LCPT was similar to that of PR-Tac, as was the short-term improvement in renal graft function. Treatment discontinuations occurred in two (6.1%) and three (8.3%) patients in the LCPT and PR-Tac groups, respectively. One patient in the LCPT group died because of a TEAE (unrelated; myocardial infarction). Acute graft rejection was observed in one patient (3.0%; T-cell-mediated rejection on day 10) in the LCPT group and in three patients (8.3%; T-cell-mediated rejection on day 12, antibody-mediated rejection on day 10, borderline rejection on day 15) in the PR-Tac group; all resolved within 11–19 days.
Discussion
The present study was the first to compare the PK and safety profiles of the two prolonged-release tacrolimus formulations, LCPT and PR-Tac, in de novo kidney transplant patients.
The proportion of patients within the target trough concentration of tacrolimus was similar between both groups, and only a few patients (< 12% in each group) had below-target tacrolimus trough levels over the study period. The PK profile following LCPT administration demonstrated significantly greater relative bioavailability (~ 30%) compared with PR-Tac at day 3 in de novo kidney transplant patients, which was maintained on days 7 and 14.
Significantly lower peak-to-trough fluctuation in concentration, a longer tmax, lower Cmax and higher AUC were also shown for LCPT versus PR-Tac, resulting in a flatter PK profile. The enhanced relative bioavailability of LCPT was accompanied by a reduction in daily dose; by day 28, the mean daily dose in the LCPT group was approximately 40% lower than that of the PR-Tac group.
These findings are consistent with those of Nigro and colleagues [10], who demonstrated sustained release of tacrolimus with a broad absorption along the gastrointestinal tract in healthy volunteers over 24 h and PK parameters consistent with the present study.
The ASERTAA study [18] compared LCPT with IR-Tac in stable African American kidney transplant recipients and reported a similar PK profile for LCPT to the results described here. The ASTCOFF study compared the PK profiles of LCPT, PR-Tac and IR-Tac and reported increased relative bioavailability and tmax for LCPT, with significantly lower Cmax achieved after post-conversion exposure normalization [9]. In addition, LCPT exhibited a similar safety profile and drug tolerability to PR-Tac, consistent with the results here and those reported previously [9, 22]. Importantly, ASERTAA and ASTCOFF investigated the PK and safety of patients switching to LCPT, thereby providing dose conversion strategies between the different formulations of tacrolimus. In contrast, the present study investigated these end points in patients initiating immunosuppressant therapy with LCPT or PR-Tac, therefore providing valuable insights not only into PK and safety, but also into dosage titration guidance for white, de novo kidney transplant patients. Notably, until the current study the PK of PR-Tac in de novo kidney transplant recipients was not well characterized, with discordant results among different studies [19].
The clinical implications of the PK characteristics of LCPT are potentially beneficial beyond reducing the risks of over- and under-dosing. Although further research is necessary, it is conceivable that reducing the variation between tacrolimus peak and trough blood concentrations may help reduce the likelihood of drug toxicity, which to date has been linked with higher AUC and trough levels [23]. Additionally, enabling once-daily administration of tacrolimus can relieve the pill burden for patients [2] and may improve patient adherence [24]. Reducing the required dose and hence the potential for side effects may also improve patient adherence [22, 25], which in turn reduces the risk of graft rejection [26, 27]. The more distal distribution of tacrolimus with LCPT may mitigate the impact of some drug-drug and drug-food interactions. Of note, tacrolimus-induced tremor may be associated with higher peak blood concentrations of tacrolimus (Cmax). The STRATO study demonstrated a significant improvement in tremor in renal transplant recipients with tacrolimus-induced hand tremors who were switched from twice-daily tacrolimus to LCPT while maintaining similar tacrolimus exposure. The impact of improved tremor potentially extends into daily activities, quality of life and psychosocial benefits for the patient [22]. Although evidence is not conclusive, an improved renal blood flow was also seen in healthy volunteers receiving a lower, once-daily Cmax formulation of tacrolimus compared with tacrolimus twice daily [28].
While a robust design was utilized, it must be emphasized that due to the exploratory nature of the study, the results are not confirmatory. The number of patients included was fairly low, although sufficient for pharmacokinetic evaluation of the tacrolimus formulations. The sample size of patients for other important factors, such as genotype, were small and may have precluded meaningful comparisons between subgroups, for example, n = 6 and n = 4 patients with CYP3A5 1*/3* genotype for LCPT and PR-Tac, respectively [29, 30]. One possible reason for this could have been that the study population was largely comprised of white males: only 5–15% of this population carry the wild-type (CYP3A5 *1) allele compared with 45–73% of African Americans [31]. In addition, investigators and patients were not blinded to the study drugs, which may be a source of unintentional bias in the evaluation of clinical outcomes and AEs, although PK data cannot be biased by unblinding and CYP3A5 polymorphism data were revealed after study end. Additionally, the short duration of the study cannot allow evaluation of the long-term evolution of the outcomes and renal function of the patients. Likewise, it must be considered that patients were all at low-standard immunologic risk, being recipients of kidneys from HLA- and ABO-compatible donors. Finally, our results may not be reproducible in kidney recipients under different immunosuppressive regimens, such as inductive lymphocyte depletion and steroid avoidance or withdrawal protocols, or in kidney recipients with extreme BMIs (< 15 or > 35 kg/m2).
Conclusion
The present study demonstrated that prolonged-release formulations of tacrolimus can reach therapeutic concentrations in the immediate post-transplant period. LCPT showed a unique PK profile in white, de novo kidney transplant recipients with greater relative bioavailability and less peak-to-trough fluctuation in blood concentrations compared with PR-Tac, longer tmax and lower Cmax. The safety profile and drug tolerability were similar between treatment groups, consistent with studies in stable kidney transplants converting their therapy to LCPT.
References
Hart A, Smith JM, Skeans MA, Gustafson SK, Wilk AR, Robinson A, et al. OPTN/SRTR 2016 annual data report: kidney. Am J Transplant. 2018;18(Suppl 1):18–113.
Kidney Disease: Improving Global Outcomes Transplant Work Group. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. 2009;9(Suppl 3):S1–155.
Food and Drug Administration. Draft guidance on tacrolimus june 2016.
Johnston A. Equivalence and interchangeability of narrow therapeutic index drugs in organ transplantation. Eur J Hosp Pharm Sci Pract. 2013;20(5):302–7.
Krzyzowska K, Kolonko A, Giza P, Chudek J, Wiecek A. No significant influence of reduced initial tacrolimus dose on risk of underdosing and early graft function in older and overweight kidney transplant recipients. Transpl Proc. 2018;50(6):1755–9.
Barraclough KAIN, Johnson DW, Campbell SB, Staatz CE. Once- versus twice-daily tacrolimus: are the formulations truly equivalent? Drugs. 2011;71(12):1561–77.
Kramer BK, Charpentier B, Backman L, Silva HT Jr, Mondragon-Ramirez G, Cassuto-Viguier E, et al. Tacrolimus once daily (ADVAGRAF) versus twice daily (PROGRAF) in de novo renal transplantation: a randomized phase III study. Am J Transplant. 2010;10(12):2632–43.
Envarsus. Summary of product characteristics. https://www.medicines.org.uk/emc/product/3670/smpc.
Tremblay S, Nigro V, Weinberg J, Woodle ES, Alloway RR. A steady-state head-to-head pharmacokinetic comparison of all FK-506 (Tacrolimus) formulations (ASTCOFF): an open-label, prospective, randomized, two-arm. Three-period crossover study. Am J Transpl. 2017;17(2):432–42.
Nigro V, Glicklich A, Weinberg J. Improved bioavailability of MELTDOSE once-daily formulation of tacrolimus (LCP-Tac) with controlled agglomeration allows for consistent absorption over 24 hrs: a scintigraphic and pharmacokinetic evaluation. Am J Transpl. 2013;13:(suppl 5):339.
Budde K, Bunnapradist S, Grinyo JM, Ciechanowski K, Denny JE, Silva HT, et al. Novel once-daily extended-release tacrolimus (LCPT) versus twice-daily tacrolimus in de novo kidney transplants: one-year results of Phase III, double-blind, randomized trial. Am J Transplant. 2014;14(12):2796–806.
Bunnapradist S, Ciechanowski K, West-Thielke P, Mulgaonkar S, Rostaing L, Vasudev B, et al. Conversion from twice-daily tacrolimus to once-daily extended release tacrolimus (LCPT): the phase III randomized MELT trial. Am J Transplant. 2013;13(3):760–9.
Rostaing L, Bunnapradist S, Grinyó JM, Ciechanowski K, Denny JE, Silva HT, et al. Novel once-daily extended-release tacrolimus versus twice-daily tacrolimus in de novo kidney transplant recipients: two-year results of phase 3, double-blind, randomized trial. Am J Kidney Dis. 2016;67(4):648–59.
Grinyo JM, Petruzzelli S. Once-daily LCP-Tacro MeltDose tacrolimus for the prophylaxis of organ rejection in kidney and liver transplantations. Expert Rev Clin Immunol. 2014;10(12):1567–79.
Staatz CE, Tett SE. Clinical pharmacokinetics of once-daily tacrolimus in solid-organ transplant patients. Clin Pharmacokinet. 2015;54(10):993–1025.
Stratta P, Quaglia M, Cena T, Antoniotti R, Fenoglio R, Menegotto A, et al. The interactions of age, sex, body mass index, genetics, and steroid weight-based doses on tacrolimus dosing requirement after adult kidney transplantation. Eur J Clin Pharmacol. 2012;68(5):671–80.
Birdwell KA, Decker B, Barbarino JM, Peterson JF, Stein CM, Sadee W, et al. Clinical pharmacogenetics implementation consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin Pharmacol Ther. 2015;98(1):19–24.
Trofe-Clark J, Brennan DC, West-Thielke P, Milone MC, Lim MA, Neubauer R, et al. Results of ASERTAA, a randomized prospective crossover pharmacogenetic study of immediate-release versus extended-release tacrolimus in african american kidney transplant recipients. Am J Kidney Dis. 2018;71(3):315–26.
Caillard S, Moulin B, Buron F, Mariat C, Audard V, Grimbert P, et al. Advagraf((R)), a once-daily prolonged release tacrolimus formulation, in kidney transplantation: literature review and guidelines from a panel of experts. Transpl Int. 2016;29(8):860–9.
Advagraf. Summary of product characteristics. https://www.medicines.org.uk/emc/product/345/smpc.
Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604–12.
Langone A, Steinberg SM, Gedaly R, Chan LK, Shah T, Sethi KD, et al. Switching STudy of Kidney TRansplant PAtients with Tremor to LCP-TacrO (STRATO): an open-label, multicenter, prospective phase 3b study. Clin Transpl. 2015;29(9):796–805.
Endrenyi L, Tothfalusi L. Metrics for the evaluation of bioequivalence of modified-release formulations. AAPS J. 2012;14(4):813–9.
Srivastava K, Arora A, Kataria A, Cappelleri JC, Sadosky A, Peterson AM. Impact of reducing dosing frequency on adherence to oral therapies: a literature review and meta-analysis. Patient Prefer Adherence. 2013;7:419–34.
Kugler C, Geyer S, Gottlieb J, Simon A, Haverich A, Dracup K. Symptom experience after solid organ transplantation. J Psychosom Res. 2009;66(2):101–10.
Morrissey PE, Reinert S, Yango A, Gautam A, Monaco A, Gohh R. Factors contributing to acute rejection in renal transplantation: the role of noncompliance. Transpl Proc. 2005;37(5):2044–7.
Tielen M, van Exel J, Laging M, Beck DK, Khemai R, van Gelder T, et al. Attitudes to medication after kidney transplantation and their association with medication adherence and graft survival: a 2-year follow-up study. J Transpl. 2014;2014:675301.
Zaltzman JS, Lai V, Schulz MZ, Moon KH, Cherney DZ. A randomized cross-over comparison of short-term exposure of once-daily extended release tacrolimus and twice-daily tacrolimus on renal function in healthy volunteers. Transpl Int. 2014;27(12):1294–302.
Lamba J, Hebert JM, Schuetz EG, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for CYP3A5. Pharmacogenet Genomics. 2012;22(7):555–8.
Lunde I, Bremer S, Midtvedt K, Mohebi B, Dahl M, Bergan S, et al. The influence of CYP3A, PPARA, and POR genetic variants on the pharmacokinetics of tacrolimus and cyclosporine in renal transplant recipients. Eur J Clin Pharmacol. 2014;70(6):685–93.
Lamba JK, Lin YS, Schuetz EG, Thummel KE. Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev. 2002;54(10):1271–94.
Acknowledgements
A special thank you to all the study participants who made this study possible.
Funding
This study and the article processing charges were funded by Chiesi Farmaceutici S.p.A., Parma, Italy. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing and Editorial Assistance
The authors received editorial/writing support for the preparation of this manuscript provided by Jennifer Mayes of Fishawack Communications, Ltd., funded by Chiesi.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole and have given their approval for this version to be published.
Disclosures
Nassim Kamar has received honoraria for lectures, consulting fees and travel grants from AbbVie, Alexion, Amgen, Astellas, Chiesi, Merck, Neovii, Novartis and Gilead. Elisabeth Cassuto has received honoraria for consulting fees and travel grants from Amgen, Astellas, Chiesi, Novartis and Roche. Giovanni Piotti received consulting fees from Chiesi for work as a medical research physician for the present study. Mirco Govoni is an employee of Chiesi Farmaceutici S.p.A. Giorgia Ciurlia is an employee of Chiesi Farmaceutici S.p.A. Silvia Geraci is an employee of Chiesi Farmaceutici S.p.A. Gianluigi Poli is an employee of Chiesi Farmaceutici S.p.A. Gabriele Nicolini is an employee of Chiesi Farmaceutici S.p.A. Christophe Mariat has received honoraria for lectures, consulting fees and travel grants from Chiesi, Novartis, Astellas and Roche. Marie Essig has received honoraria for lectures, consulting fees and travel grants from Astellas, Chiesi, Novartis and Gilead. Paolo Malvezzi has received honoraria for lectures and travel grants from Astellas, Chiesi, Neovii and Sanofi. Yannick Le Meur has received honoraria for lectures or consulting fees from Astellas, BMS, Chiesi, Novartis, Otsuka, Roche and Sanofi. Lionel Rostaing received honoraria for lectures and travel grants from Chiesi, Novartis, AbbVie, Amgen, Hikma, Neovii and Sanofi. Valeria Garrigue and Arnaud Del Bello have nothing to disclose.
Compliance with Ethics Guidelines
All procedures performed in studies involving human participants were according to the clinical study protocol, the current International Council for Harmonization Good Clinical Practice guidelines, all local guidelines and the Declaration of Helsinki (1964 and amendments). The independent ethics committee/institutional research board for this study was the Committee for the Protection of Persons South Mediterranean V, CHU de Nice-Hôpital De Cimiez. Informed consent was obtained from all individual participants included in the study. A specific Patient Information Sheet-Informed Consent Form for the genotyping test was obtained.
Data Availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced digital features
To view enhanced digital features for this article go to: https://doi.org/10.6084/m9.figshare.7392524.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kamar, N., Cassuto, E., Piotti, G. et al. Pharmacokinetics of Prolonged-Release Once-Daily Formulations of Tacrolimus in De Novo Kidney Transplant Recipients: A Randomized, Parallel-Group, Open-Label, Multicenter Study. Adv Ther 36, 462–477 (2019). https://doi.org/10.1007/s12325-018-0855-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-018-0855-1