
Abstract
An endolymphatic sac tumor (ELST) is a rare, indolent but locally aggressive tumor arising in the posterior petrous ridge. Patients present with sensorineural hearing loss and tinnitus. As the tumor progresses, patients may experience vertigo, ataxia, facial nerve paresis, pain and otorrhea. Most patients present in their 4th or 5th decade with a wide age range. Patients with von Hippel–Lindau disease have an increased likelihood of developing ELST. Histologically, ELST is a low-grade adenocarcinoma. As it progresses, it destroys bone and extends into adjacent tissues. The likelihood of regional or distant metastases is remote. The optimal treatment is resection with negative margins. Patients with positive margins, gross residual disease, or unresectable tumor are treated with radiotherapy or radiosurgery. Late recurrences are common, so long follow-up is necessary to assess efficacy. The likelihood of cure depends on tumor extent and is probably in the range of 50–75%.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Like other parts of the membranous labyrinth, the endolymphatic sac is derived from the ectodermal otocyst and is located on the posteromedial aspect of the temporal bone [1, 2]. The proximal segment of the endolymphatic sac is contiguous with the endolymphatic duct [3]. The posterior segment of the endolymphatic sac is in the dura of the posterior cranial fossa. While the main functions of the sac relate to monitoring the volume and pressure of endolymph, as well as eliminating waste products therein via heterophagy, an endocrine/paracrine function has recently been considered [4, 5]. Different kinds of failure in the endolymphatic sac homeostasis have been implicated in the development of Ménière’s disease [6].
In 1988, Gaffey et al. described a locally destructive tumor of the temporal bone in the region of the endolymphatic sac [7]. The authors reviewed 9 similar cases identified retrospectively in the English literature and suggested the term “aggressive papillary middle ear tumor” for an entity distinct from middle ear adenoma [7]. The following year, Heffner reported 20 similar cases, referring to them as low-grade adenocarcinoma of probable endolymphatic sac origin [8]. The terminology has been reviewed by Mills et al., but eventually the WHO endorsed the widely popular term ‘endolymphatic sac tumor’ (ELST) [9]. It should be, however, noted that a definite origin from the endolymphatic sac remains to be established.
ELSTs are rare, with fewer than 300 cases reported in the literature [10,11,12,13]. The aim of this article is to review the presentation, diagnosis, treatment, and outcomes for this entity. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Clinical and Radiographic Features
Patients with ELST often present with the gradual onset of sensorineural hearing loss and tinnitus [14,15,16,17,18,19,20,21,22]. This may slowly progress to include earache, vertigo, ataxia, facial nerve paresis, and otorrhea (Table 1) [14]. Most patients present in their 30s or 40s with a wide age range [23, 24]. Husseini et al. analyzed 107 patients who were treated by the Gruppo Otologico (Piacenza, Italy) or reported in the literature (only papers including 3 or more patients were considered) and observed the following presenting symptoms: hearing loss, 94%; tinnitus, 55%; vertigo, 47%; facial palsy, 33%; lower cranial nerve deficits, 5%; and facial paresthesias, 5% [11]. Thus, particularly in patients with clinical manifestations simulating Ménière’s disease, the endolymphatic sac region must be carefully examined in order not to miss an ELST [25].
ELSTs may be sporadic or associated with von Hippel–Lindau (VHL) disease which is attributable to inactivation of a tumor suppressor gene on the short arm of chromosome 3, and inherited as an autosomal dominant trait with variable expression [1, 14, 15, 20, 26,27,28,29,30,31,32,33,34,35,36,37]. VHL predisposes patients to multiple hemangioblastomas of the retina or central nervous system as well as tumors and cysts in various organs, including renal cell carcinoma, pheochromocytoma, paraganglioma, ELST, epididymal cystadenoma, and pancreatic serous cystadenoma or neuroendocrine tumors [1]. The incidence of ELSTs in patients with VHL may be as high as 24% [1], but this is not universally accepted. Bausch et al. reported on 1789 patients with VHL who were evaluated in all university and large city hospitals with otolaryngology units in Germany, France, and The Netherlands, as well as selected centers in Spain and Italy [27]. Among the 93 ELST registrants, 52 (56%) presented with the characteristic clinical features of VHL disease. Although the remaining 41 registrants (44%) presented with apparently sporadic ELST, 16 (39%) among these were found to harbor a VHL germline mutation. Thus, 25 patients had truly sporadic ELSTs and 68 had VHL-associated ELSTs, so that the incidence of ELSTs in VHL was 3.6% in the entire series. When the two groups were compared, patients with sporadic ELST tended to be older, with a mean age of 40 years (range 12–78) versus 30 years (range 6–62), less likely to be bilateral (0 vs. 9%), and more likely to be male (60 vs. 38%). ELST was observed at initial presentation of VHL in 32% of the patients. The fact that ELST can be the first manifestation of VHL disease underscores the importance of early diagnosis of VHL-associated ELSTs, preferably with VHL germline mutation analysis in all apparently sporadic ELST cases [27]. Conversely, all patients with VHL should receive regular clinical screening for ELST. This is important in order to perform a complete tumor resection preserving hearing function, which is crucial in these patients, who often suffer from vision loss and gait disturbances associated with VHL.
As ELST progresses, it may extend medially into the cerebellopontine angle, superiorly to the middle cranial fossa, laterally into the middle ear, and anteromedially to the cavernous sinus [1]. The likelihood of regional lymph node metastases is remote [1]. The probability of distant metastases is also very low, although drop metastases in the spinal canal have been reported [26].
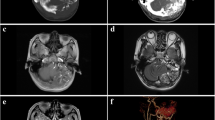
Radiographic evaluation with high-resolution computed tomography (CT) using a thin-slice bone algorithm may reveal bone destruction of the posterior aspect of the petrous ridge (Fig. 1) [38,39,40,41]. Magnetic resonance imaging (MRI) usually reveals a heterogeneous mass which is hyperintense on contrast enhanced T1 and T2 sequences due to hypervascularity [38, 42]. The heterogeneous appearance may be due to blood products, cysts, cholesterol crystals, calcifications, and flow voids due to vessels [38]. Digital subtraction angiography is helpful for displaying the tumor’s vascular structure and supply, which is often complex and may help to map pre-surgical landmarks and to differentiate ELST from other lesions [43]. Increased activity on the Ga 68-DOTATATE positron emission tomography (PET/CT), used to search for VHL-associated neuroendocrine tumors, has also been reported in ELST, indicating cell-surface expression of somatostatin receptors by this tumor [26].

a, b MRI showing a mass that is hyperintense tumor-like mass (asterisk) on contrast enhanced T1 sequence. c, d CT showing bone destruction of the posterior aspect of the petrous ridge. Arrows point to the tumor
Pathologic Features
ELSTs are a low-grade adenocarcinoma [8, 9]. They may be very vascular and biopsy may be complicated by bleeding, which may be severe [32]. In addition, the origin of the tumor in the region of the inner ear makes it inaccessible for a biopsy unless this is intra-operative. Macroscopically, the tumor is red/brown and fibrous.
Histologically, various growth patterns of papillary, solid, follicular or cystic architecture are seen. Papillary or luminal arrangements usually show a single layer of tumor cells (Fig. 2a, b). The tumor cells may be flattened, cuboidal or columnar with bland central or excentrically-located nuclei and pale eosinophilic or clear cytoplasm (Fig. 2b, c). Cellular pleomorphism, mitoses and necrosis are not seen. Small glands and follicular structures containing deeply eosinophilic colloid-like and periodic acid–Schiff-positive material combined with papillary arrangements simulate thyroid tumors. However, the nuclear features characteristic of papillary thyroid carcinoma are not a feature of ELST. The vascularity of the papillary arrangements bears a resemblance to choroid plexus papillomas.

a Hematoxylin and eosin-stained section (×100 magnification). Microscopy of an endolymphatic sac tumor shows dilated epithelial cell-lined follicle-like cystic structures with secretions. The cells are uniform with small hyperchromatic nuclei. b Hematoxylin and eosin-stained section (×200 magnification). At higher magnification, the uniformity of the endolymphatic sac tumor cells are noted with round nuclei and no mitoses. The architectural pattern is undulating with papillary formations. c Hematoxylin and eosin-stained section (×200 magnification). The enlargement and clearing of the cytoplasm in this endolymphatic sac tumor mimics renal cell carcinoma. The tumor here is noted to be vascular and adjacent to bone (top). This tumor must be differentiated from renal cell carcinoma which also occurs in VHL patients. d Immunohistochemical evaluation for pancytokeratin (×200 magnification) highlights the epithelial cells lining the papillary structures in this endolymphatic sac tumor
Immunohistochemically, ELST stains for pan-cytokeratins as well as cytokeratins CK5, CK7 and CK19, but CK20 is negative (Fig. 2d). There is variable expression of epithelial membrane antigen, Ber EP4, glial fibrillar acid protein, and S-100 protein. There is no expression of thyroglobulin or transcription termination factor 1. The proliferation rate as assessed by the Ki67 immunostain is low.
The ultrastructure of ELST has been briefly addressed by Heffner [8]. Electron microscopic examination shows a single epithelial layer overlying the basal lamina. The cells show short interdigitating cytoplasmic processes joined by desmosomes. Apical microvilli are present with moderate amounts of intracytoplasmic organelles. Occasional electron-dense neurosecretory-type granules are identified that are 200–350 μm in diameter.
The differential diagnosis includes middle ear adenoma, middle ear adenocarcinoma, jugulotympanic paraganglioma, choroid plexus papilloma, non-specific ceruminal gland adenocarcinoma, and metastatic adenocarcinoma (particularly thyroid and renal primaries) which typically show greater nuclear irregularities [14, 38, 44,45,46]. Diaz et al. reported on 134 temporal bone lesions evaluated at the University of California-Davis between 1994 and 2005: paragangliomas, 78%; jugular foremen schwannomas, 4%; hemangiomas 4%; 7th nerve schwannomas, 4%; endolymphatic sac lesions, 4%; metastases, 3%; middle ear adenoma, 1%; and chondrosarcoma, 1% [43]. Of the endolymphatic sac lesions, 3 were ELSTs and 2 were pseudotumors. Thus, 3 of 134 lesions (2%) were ESLTs [43].
Staging
There are no universally accepted staging recommendations. Bambakidis et al. suggested the following system: stage I, tumor confined to the temporal bone and middle ear cavity; stage II, extension to the posterior fossa; stage III, extension to the middle cranial fossa; and stage IV, extension to the clivus and/or sphenoid wing [13].
The later system by Schipper et al. distinguishes type-A, locally confined, tumors without temporal bone erosion or infiltration of the dura; type-B tumors showing evidence of infiltration of the osseous labyrinth and sensorineural hearing loss; and type-C tumors further invading the sigmoid sinus and jugular bulb [47].
These systems are revisited below in conjunction with treatment.
Treatment
The majority of patients reported in the literature were treated surgically (Fig. 3). Experience with stereotactic radiosurgery (SRS) and with fractionated radiotherapy (RT) is limited [3, 8, 48, 49]. Generally, low-grade adenocarcinomas are unlikely to be radiosensitive and complete resection with negative margins is likely to be the treatment of choice.

a 1 Tumor is located under a bulging dura; 2 VIII cranial nerve; 3 lower cranial nerves. b 1 Operative cavity in the posterior aspect of the petrous ridge; 2 VIII cranial nerve; arrows indicate the dural resection margins
SURGICAL TREATMENT
ELSTs are sometimes confined to the endolymphatic sac and duct, but more often erode the petrous bone or the mastoid air cells around the endolymphatic sac, including the posterior semicircular canal. However, larger tumors extend medially and inferiorly via the retrofacial route to invade the jugular bulb and the hypotympanum, and rarely invade the sigmoid sinus. Larger tumors often involve the posterior fossa dura and extend posteriorly to the cerebellopontine angle and anteriorly to the internal auditory canal, occasionally reaching the middle fossa across the tegmen mastoideum [50]. These pathways of invasion have to be considered when planning the surgical approach.
According to Nevoux et al. [29], there are two types of tumors. The first type infiltrates the bone and adjacent structures and is composed of fibrous tissue, but it is not markedly hemorrhagic. The second type is a cystic tumor form, which is weakly infiltrative and heavily hemorrhagic. Large hypervascularized cystic tumors with significant extension into posterior fossa may benefit from preoperative embolization before surgical resection to minimize intraoperative bleeding and morbidities, as well as to facilitate complete surgical resection. The blood supply arises mainly from the branches of the external carotid artery (posterior auricular artery, occipital artery, and ascending pharyngeal artery). The blood supply of huge intracranial growths may also be from branches of the vertebral artery, such as the anterior inferior cerebellar artery [50, 51].
The type of approach depends on the hearing level and tumor size and localization. Sporadic tumors usually show worse hearing function and have a more delayed diagnosis. Clinical symptoms and aggressiveness of the tumor are more serious in the sporadic cases than in the cases of VHL, and the treatment should be more extensive to prevent recurrences. Conversely, in VHL-associated tumors, the goals of the treatment are to perform surgery as soon as possible with the least amount of hearing loss [29].
According to Schipper et al., type-A ELST is locally confined and therefore amenable to total ablation through a transmastoidal–retrolabyrinthine approach with the preservation of the facial and vestibulocochlear nerves and inner ear structures [47]. A mastoidectomy is performed with identification of the horizontal semicircular canal and the third segment of the facial nerve. The jugular bulb is skeletonized, as is the tegmental dura and sinodural angle. Once the posterior semicircular canal is skeletonized, the outline of a bulging tumor involving the endolymphatic sac is seen [10]. When the posterior fossa dura is healthy, the sac and duct are removed en bloc. In the case of involvement of both dural leaflets of the endolymphatic sac, the dura in Trautmann’s triangle should be resected and the procedure transformed into a retrolabyrinthine–transdural approach. This would result in a transmastoid cerebrospinal fluid leak—hence, the space must be sealed-off by duroplasty using autologous fat grafts. Preservation of hearing requires that the integrity of the perilymphatic space should not be violated by fenestrating the semicircular canals. Whenever possible, one should aim to maintain hearing, since the incidence of bilateral ELSTs can be as high as 30% in VHL [52]. Cochlear implantation has proved to be successful for aural rehabilitation in deaf patients with ELSTs and, if it is considered in bilateral ELST cases, surgical approaches should be tailored to keep the otic capsule intact in order to achieve optimal cochlear implant benefit [50].
Preservation of hearing is also allowed using a retrosigmoid approach, which permits a better control of cerebellopontine angle tumor extensions than the retrolabyrinthine approach, as well as facilitates dural resection. In contrast, the retrosigmoid approach does not afford visualization of the posterior semicircular canal during dissection of the endolymphatic duct.
A translabyrinthine approach with facial nerve preservation would be appropriate for type-B ELST, mainly in patients with poor or unserviceable hearing, who often have labyrinthine invasion [47]. Since the inner ear may already be eroded or destroyed, the lateral wall of the internal auditory canal can be drilled away. For complete removal, dural excision and duroaplasty are also required. Alternatively, a retrosigmoid approach may be considered.
For tumors with deeper involvement of the petrous bone, a transcochlear approach can be needed, when complete exenteration of the otic capsule and the exposure of the petrous carotid artery is necessary [53]. A combined transtemporal–retrosigmoid approach or even subtotal resection of the temporal bone gives adequate exposure for tumors involving the posterior and middle cranial fossa.
For type-C ELST, the infratemporal approach is recommended for safe distal and proximal ligation and resection of the sigmoid sinus and jugular bulb in large lesions when the tumor involves the facial nerve, the jugular bulb, and the middle ear. Approximately 10–30% of ELST patients present preoperatively with facial nerve weakness or paralysis [29, 50, 51]. The facial nerve should be preserved if the nerve is not invaded. This implies a total transposition of the facial nerve (Fisch A infratemporal fossa approach) or, preferably, the use of the fallopian bridge technique which preserves the integrity and vascularity of the facial nerve [47, 54, 55]. In nearly 50% of the patients, the posterior wall of the external auditory canal must be demolished for better exposure of the carotid artery, and the middle ear cleft is obliterated after total removal of the tumor [55]. Otherwise, involved facial nerves are removed, and the great auricular nerve is used for neural transplantation, or a facial nerve–hypoglossal nerve anastomosis is performed in the case of invasion of the proximal stump of the nerve. This should be followed by complete removal of the tumor [47, 54].
According to the staging system proposed by Bambakidis et al., stage I and II ELSTs can be removed by transmastoid, translabyrinthine, retrolabyrinthine or retrosigmoid approaches depending on the extension to the middle ear and the presence of sensorineural hearing loss [13]. Stage III and IV lesions require more complex approaches depending on the structures involved (subtemporal craniotomy with petrosectomy, modified transcochlear approaches, staged anterior and posterior fossa techniques, etc.) [13, 56].
RADIATION THERAPY TREATMENT
External-beam RT should be considered for patients with close or positive margins as well as those with gross disease, either after subtotal resection or in patients considered inoperable due to morbidity or with tumors deemed incompletely resectable due to anatomical conditions, (e.g., cranial nerve involvement, complex vascular supply) [43]. Because the likelihood of metastases is low, only the primary site is irradiated. Given the behavior of ELST as a low-grade adenocarcinoma, “carcinoma doses” should be employed so that patients with close (< 5 mm) or positive margins receive 66 Gy in 33 fractions and patients with gross disease receive 70 Gy in 35 fractions or their radiobiological dose equivalents. The difficulties in suggesting RT dose recommendations are influenced by limited data and unstated RT dose or dose-fractionation schedules.
Intensity modulated radiotherapy should be employed if patients are treated with photons due to the superior dose distribution in comparison to other techniques. Proton beam irradiation may be used rather than photon-based RT, if available, to reduce the dose to the organs at risk and hopefully reduce the likelihood of late complications [57].
SRS may be employed alone or combined with external beam RT to treat areas of incompletely resected disease that are approximately 3 cm or less in maximum diameter. SRS may be delivered with a Gamma Knife, a linear accelerator-based system, or proton beam. The optimal marginal dose for SRS is probably 15–18 Gy. Sporadic but encouraging experiences on the use of SRS for post-surgical recurrences of ELST have been reported in the literature, although the risk of a marginal miss after SRS alone should not be underestimated [58, 59].
Outcomes
Evaluation of outcomes is hampered by the rarity of ELST, the limited number of patients included in reports, variable treatment, and short follow-up. Patel et al. reported on a patient treated with RT for presumed temporal bone paraganglioma who, 13 years later, was found to have an ELST, highlighting the necessity for long follow-up to assess the efficacy of treatment [60]. That said, the following are some institutional experiences and a recent literature review.
Nevoux et al. reported on 6 sporadic and 8 VHL-related ELSTs treated at two tertiary care centers [29]. Two patients with small tumors were observed; they died at 6 years of metastatic renal carcinoma (1 patient) and gastric cancer (1 patient). The remaining patients were treated surgically. Four of 6 sporadic tumors and 5 of 6 VHL tumors were locally controlled—hence, the overall local control rate was 9 of 12 (75%). Follow-up ranged from 0.5 to 10 years (median 4 years); 1 patient had no follow-up.
Rodrigues et al. reported on 7 patients treated surgically at St. Vincent’s Hospital (Sydney): 1 was alive with disease at 63 months; 1 patient died of intercurrent disease at 120 months; and 5 patients were alive and disease-free at 6, 10, 41, 98, and 144 months [61].
Carlson et al. reported on 13 patients with unilateral (12) or bilateral (1) ELSTs treated at the Mayo Clinic between 1988 and 2010 [58]. Sporadic tumors were seen in 8 patients and associated with VHL in 5 patients. Twelve patients were previously untreated and 11 were treated surgically; 10 were controlled and 1 developed a local recurrence successfully salvaged with a second operation. Follow-up ranged from 5 to 121 months (median 76 months); 6 patients were followed for more than 5 years. A single infirm patient was subjected to SRS with a marginal dose of 15 Gy and remained locally controlled at 94 months. Two patients were operated for a local recurrence after surgery elsewhere; one patient was locally controlled at 2 years, and 1 patient experienced additional recurrence which was locally controlled 11 months after SRS.
Kim et al. [50] reported on 31 patients, including 4 with bilateral ELSTs, who underwent resection of 33 ELSTs at the NIH (Bethesda). Complete tumor resection was achieved in 30 ears (91%). The tumor remained stable in 1 patient (follow-up 78 months) but progressed in the other (follow-up 105 months). Two of these patients underwent focused irradiation. There was only 1 case of recurrence after initial complete gross resection (46 months postresection) which was successfully resected.
Friedman et al. reported on 18 patients treated at the House Ear Institute (Los Angeles) between 1971 and 2011 and followed from 1 to 261 months (mean 67 months) [18]. Fifteen of 18 patients (83%) treated surgically remained locally controlled, including 1 patient who also received RT to 70 Gy. One patient was operated on 3 times plus 48 Gy and was alive with disease at 261 months, 1 patient was operated on 4 times plus 54 Gy and died with disease at 18 months, and 1 patient who underwent surgery alone was alive with disease at 88 months. Six patients who were locally controlled had follow-up of less than 1 year, including 3 who were lost to follow-up. Excluding these 6 patients, the local control of the remaining 12 patients with 1 or more years of follow-up was 9/12 (75%).
Heffner reported a clinicopathologic study of 20 patients from the Armed Forces Institute of Pathology [8]. One patient with inadequate data was excluded. Seven patients underwent a subtotal resection and 1 of 7 died postoperatively. Four patients underwent postoperative RT to 47.15, 47.15, and 50.29 Gy. The RT dose was not specified for 1 patient. Five patients developed a local recurrence from 1 to 6 years following surgery (mean 2.4 years). Two patients subsequently underwent a gross total resection and were disease-free at 2 and 8 years, respectively. Twelve patients initially underwent a gross total resection and 11 of 12 patients (92%) remained disease-free from 2 to 12 years after surgery. The remaining patient developed a questionable local recurrence at 2 years.
Kunzel et al. reported on 1 patient with a sporadic 3.5-cm ELST who underwent resection with an R1 margin followed by postoperative RT to 60 Gy [16]. The patient remained disease-free at 10 years [11].
Balasubramaniam et al. reported on a patient who had a local recurrence after surgery who was treated with SRS to a marginal dose of 15 Gy and remained disease-free at 2.5 years [48].
On the other hand, Hou et al. reported on 11 patients with ELST; 9 of 11 underwent a complete resection and all were disease-free after a follow-up period of 14 months to 10 years [51].
Husseini conducted a literature search that encompassed 16 publications with 3 or more patients, including a total of 107 patients of whom 106 patients underwent surgery [11]. Subtotal resection was performed in 11%, 19% underwent preoperative embolization, and 22% received postoperative RT or SRS. Follow-up ranged from 1 month to 21 years. Five percent of patients were dead with disease within 2 years and 10% were alive with disease. Tumor recurred in 10% of patients within 8 months to 13 years after treatment.
Conclusion
ELST is a rare, slow growing, but locally destructive, temporal bone low-grade adenocarcinoma that may be sporadic or associated with VHL. The likelihood of regional and distant metastases is low. If uncontrolled, ELST may cause death by local disease progression. Wide excision with negative margins is the treatment of choice. Patients with positive margins, incompletely resected gross disease or inoperable tumors should be considered for RT or SRS.
References
Zanoletti E, Girasoli L, Borsetto D, et al. Endolymphatic sac tumour in von Hippel–Lindau disease: management strategies. Acta Otorhinolaryngol Ital. 2017;37:423–9.
Batsakis JG, el-Naggar AK. Papillary neoplasms (Heffner’s tumors) of the endolymphatic sac. Ann Otol Rhinol Laryngol. 1993;102:648–51.
Poletti AM, Dubey SP, Colombo G, et al. Treatment of endolymphatic sac tumour (Papillary adenocarcinoma) of the temporal bone. Rep Pract Oncol Radiother. 2016;21:391–4.
Moller MN, Kirkeby S, Vikesa J, et al. The human endolymphatic sac expresses natriuretic peptides. Laryngoscope. 2017;127:E201–8.
Couloigner V, Teixeira M, Sterkers O, et al. The endolymphatic sac: its roles in the inner ear. Med Sci (Paris). 2004;20:304–10.
Mori N, Miyashita T, Inamoto R, et al. Ion transport its regulation in the endolymphatic sac: suggestions for clinical aspects of Meniere’s disease. Eur Arch Otorhinolaryngol. 2017;274:1813–20.
Gaffey MJ, Mills SE, Fechner RE, et al. Aggressive papillary middle-ear tumor. A clinicopathologic entity distinct from middle-ear adenoma. Am J Surg Pathol. 1988;12:790–7.
Heffner DK. Low-grade adenocarcinoma of probable endolymphatic sac origin A clinicopathologic study of 20 cases. Cancer. 1989;64:2292–302.
Mills S, Stelow E, Hunt J. Aggressive papillary tumor of temporal bone and endolymphatic sac. In: Tumors of upper aerodigestive tract and ear. AFIP Atlas of Tumor Pathology. Silver Spring: American Registry of Pathology;2012.
Wick CC, Manzoor NF, Semaan MT, et al. Endolymphatic sac tumors. Otolaryngol Clin North Am. 2015;48:317–30.
Husseini ST, Piccirillo E, Taibah A, et al. The Gruppo Otologico experience of endolymphatic sac tumor. Auris Nasus Larynx. 2013;40:25–31.
Megerian CA, Semaan MT. Evaluation and management of endolymphatic sac and duct tumors. Otolaryngol Clin North Am. 2007;40:463–78, viii.
Bambakidis NC, Megerian CA, Ratcheson RA. Differential grading of endolymphatic sac tumor extension by virtue of von Hippel–Lindau disease status. Otol Neurotol. 2004;25:773–81.
Lucas C, Leclère JC, Mornet E, et al. Intralabyrinthine sporadic endolymphatic sac tumour. Eur Ann Otorhinolaryngol Head Neck Dis. 2018;135:123–5.
Schnack DT, Kiss K, Hansen S, et al. Sporadic endolymphatic sac tumor-A very rare cause of hearing loss, tinnitus, and dizziness. J Int Adv Otol. 2017;13:289–91.
Kunzel J, Agaimy A, Hornung J, et al. Sporadic endolymphatic sac tumor–a diagnostic and therapeutic challenge. Int J Clin Exp Pathol. 2014;7:2641–6.
Li JC, Brackmann DE, Lo WW, et al. Reclassification of aggressive adenomatous mastoid neoplasms as endolymphatic sac tumors. Laryngoscope. 1993;103:1342–8.
Friedman RA, Hoa M, Brackmann DE. Surgical management of endolymphatic sac tumors. J Neurol Surg B. 2013;74:12–9.
Butman JA, Nduom E, Kim HJ, et al. Imaging detection of endolymphatic sac tumor-associated hydrops. J Neurosurg. 2013;119:406–11.
Biswas R, Kumari K, Gupta S, et al. Endolymphatic sac tumor in von Hippel-Lindau disease: a rare case report. J Pediatr Neurosci. 2017;12:294–7.
Zulkarnaen M, Tang IP, Wong SL. Endolymphatic sac tumour. Malays J Pathol. 2012;34:53–5.
Codreanu C, Tran Ba Huy P. Isolate vertigo crisis revealing an endolymphatic sac tumor. Roman J Morphol Embryol. 2010;51:387–9.
Kupferman ME, Bigelow DC, Carpentieri DF, et al. Endolymphatic sac tumor in a 4-year-old boy. Otol Neurotol. 2004;25:782–6.
Devaney KO, Boschman CR, Willard SC, et al. Tumours of the external ear and temporal bone. Lancet Oncol. 2005;6:411–20.
Lee KJ, Kirsch CF, Lai C, et al. Endolymphatic sac tumor presenting with Meniere’s disease. Otolaryngol Head Neck Surg. 2010;142:915–6.
Papadakis GZ, Millo C, Sadowski SM, et al. Endolymphatic sac tumor showing increased activity on 68 Ga DOTATATE PET/CT. Clin Nucl Med. 2016;41:783–4.
Bausch B, Wellner U, Peyre M, et al. Characterization of endolymphatic sac tumors and von Hippel–Lindau disease in the International Endolymphatic Sac Tumor Registry. Head Neck. 2016;38(Suppl 1):E673–9.
Du J, Wang J, Cui Y, et al. Clinicopathologic study of endolymphatic sac tumor (ELST) and differential diagnosis of papillary tumors located at the cerebellopontine angle. Neuropathology. 2015;35:410–20.
Nevoux J, Nowak C, Vellin JF, et al. Management of endolymphatic sac tumors: sporadic cases and von Hippel–Lindau disease. Otol Neurotol. 2014;35:899–904.
Bastier PL, de Mones E, Marro M, et al. Endolymphatic sac tumors: experience of three cases. Eur Arch Otorhinolaryngol. 2013;270:1551–7.
Eze N, Huber A, Schuknecht B. De novo development and progression of endolymphatic sac tumour in von Hippel-Lindau disease: an observational study and literature review. J Neurol Surg B. 2013;74:259–65.
Bellairs JA, Gluth MB. A histopathological connection between a fatal endolymphatic sac tumour and von Hippel-Lindau disease from 1960. J Laryngol Otol. 2018;132:75–8.
de Minteguiaga C, Garcia Ibanez L, Tran Ba Huy P. [Endolymphatic sac tumor and von Hippel-Lindau disease. Review of the literature]. Acta Otorrinolaringol Esp. 2002;53:515–20.
Rao Q, Zhou J, Wang JD, et al. Endolymphatic sac tumor with von Hippel-Lindau disease: report of a case with analysis of von Hippel–Lindau gene and review. Ann Diagn Pathol. 2010;14:361–4.
Yang X, Liu XS, Fang Y, et al. Endolymphatic sac tumor with von Hippel-Lindau disease: report of a case with atypical pathology of endolymphatic sac tumor. Int J Clin Exp Pathol. 2014;7:2609–14.
Dornbos D 3rd, Kim HJ, Butman JA, et al. Review of the neurological implications of von Hippel-Lindau disease. JAMA Neurol. 2018;75:620–7.
Skalova A, Sima R, Bohus P, et al. Endolymphatic sac tumor (aggressive papillary tumor of middle ear and temporal bone): report of two cases with analysis of the VHL gene. Pathol Res Pract. 2008;204:599–606.
Jegannathan D, Kathirvelu G, Mahalingam A. Three sporadic cases of endolymphatic sac tumor. Neurol India. 2016;64:1336–9.
Shaigany K, Vazquez A, Kwong KM, et al. Endolymphatic sac tumor in association with von Hippel–Lindau syndrome. Ear Nose Throat J. 2016;95:96–8.
Yilmaz I, Bolat F, Demirhan B, et al. Endolymphatic sac papillary tumor: a case report and review. Auris Nasus Larynx. 2008;35:276–81.
Ho VT, Rao VM, Doan HT, et al. Low-grade adenocarcinoma of probable endolymphatic sac origin: CT and MR appearance. Am J Neuroradiol. 1996;17:168–70.
Geerts B, Bernardeschi D, Di Maria F, et al. Post-surgery pCASL perfusion MRI of endolymphatic sac tumor. J Neuroradiol. 2017;44:345–7.
Diaz RC, Amjad EH, Sargent EW, et al. Tumors and pseudotumors of the endolymphatic sac. Skull Base. 2007;17:379–93.
Pollak A. Correspondence re: Lavoie M, Morency RM: Low-grade papillary adenomatous tumors of the temporal bone: report of two cases and review of the literature. Mod Pathol 8:603, 1995. Mod Pathol. 1996;9:460–1.
Olcott C, Strasnick B. A blue middle ear mass: Cholesterol granuloma mimicking a glomus tumor and endolymphatic sac tumor. Am J Otolaryngol. 2017;38:100–2.
Malhotra S, Rao RV, Valiathan M, et al. Low-grade adenocarcinoma of endolymphatic sac origin. Am J Otolaryngol. 2006;27:362–5.
Schipper J, Maier W, Rosahl SK, et al. Endolymphatic sac tumours: surgical management. J Otolaryngol. 2006;35:387–94.
Balasubramaniam S, Deshpande RB, Misra BK. Gamma knife radiosurgery in jugular foramen endolymphatic sac adenocarcinoma. J Clin Neurosci. 2009;16:710–1.
Ferreira MA, Feiz-Erfan I, Zabramski JM, et al. Endolymphatic sac tumor: unique features of two cases and review of the literature. Acta Neurochir (Wien). 2002;144:1047–53.
Kim HJ, Hagan M, Butman JA, et al. Surgical resection of endolymphatic sac tumors in von Hippel–Lindau disease: findings, results, and indications. Laryngoscope. 2013;123:477–83.
Hou ZH, Huang DL, Han DY, et al. Surgical treatment of endolymphatic sac tumor. Acta Otolaryngol. 2012;132:329–36.
Manski TJ, Heffner DK, Glenn GM, et al. Endolymphatic sac tumors. A source of morbid hearing loss in von Hippel–Lindau disease. JAMA. 1997;277:1461–6.
Poletti AM, Dubey SP, Barbo R, et al. Sporadic endolymphatic sac tumor: its clinical, radiological, and histological features, management, and follow-up. Head Neck. 2013;35:1043–7.
Suarez C, Sevilla MA, Llorente JL. Temporal paragangliomas. Eur Arch Otorhinolaryngol. 2007;264:719–31.
Llorente JL, Obeso S, Lopez F, et al. Comparative results of infratemporal fossa approach with or without facial nerve rerouting in jugular fossa tumors. Eur Arch Otorhinolaryngol. 2014;271:809–15.
Sanna M, Mazzoni A, Saleh E, et al. The system of the modified transcochlear approach: a lateral avenue to the central skull base. Am J Otol. 1998;19:88–97 (discussion 97-8).
Mendenhall NP, Malyapa RS, Su Z, et al. Proton therapy for head and neck cancer: rationale, potential indications, practical considerations, and current clinical evidence. Acta Oncol. 2011;50:763–71.
Carlson ML, Thom JJ, Driscoll CL, et al. Management of primary and recurrent endolymphatic sac tumors. Otol Neurotol. 2013;34:939–43.
Sinclair G, Al-Saffar Y, Brigui M, et al. Gamma knife radiosurgery in the management of endolymphatic sac tumors. Surg Neurol Int. 2018;9:18.
Patel PC, Pellitteri PK, Reams CL, et al. Aggressive papillary tumor of the temporal bone: delayed extensive recurrence following radiation therapy. Skull Base Surg. 1997;7:45–8.
Rodrigues S, Fagan P, Turner J. Endolymphatic sac tumors: a review of the St. Vincent’s hospital experience. Otol Neurotol. 2004;25:599–603.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Medical Writing and/or Editorial Assistance
We would like to thank Jessica Kirwan and the editorial staff of the University of Florida Department of Radiation Oncology for assistance in preparing the manuscript for submission and publication.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
William M. Mendenhall, Carlos Suárez, Alena Skálová, Primož Strojan, Asterios Triantafyllou, Kenneth O. Devaney, Michelle D Williams, Alessandra Rinaldo and Alfio Ferlito have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article was written by members of the International Head and Neck Scientific group (www.IHNSG.com).
Enhanced digital content
To view enhanced digital content for this article go to https://doi.org/10.6084/m9.figshare.6245666.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Mendenhall, W.M., Suárez, C., Skálová, A. et al. Current Treatment of Endolymphatic Sac Tumor of the Temporal Bone. Adv Ther 35, 887–898 (2018). https://doi.org/10.1007/s12325-018-0730-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-018-0730-0