
Abstract
Introduction
Semaglutide is a glucagon-like peptide-1 analogue for once-weekly subcutaneous treatment of type 2 diabetes. This trial compared the pharmacokinetics, pharmacodynamics, and safety of semaglutide in Japanese and Caucasian subjects.
Methods
In this single-center, double-blind, parallel-group, 13-week trial, 44 healthy male subjects (22 Japanese, 22 Caucasian) were randomized within each race to semaglutide 0.5 mg (n = 8), 1.0 mg (n = 8), placebo 0.5 mg (n = 3) or 1.0 mg (n = 3). The primary endpoint was semaglutide exposure at steady state [area under the curve (AUC0–168h)].
Results
Steady-state exposure of semaglutide was similar for both populations: AUC0–168h estimated race ratio (ERR), Japanese/Caucasian: 0.5 mg, 1.06; 1.0 mg, 0.99; maximum concentration (Cmax) ERR: 0.5 mg, 1.06; 1.0 mg, 1.02. Exposure after the first dose (0.25 mg) was slightly higher in Japanese versus Caucasian subjects (AUC0–168h ERR 1.11; Cmax ERR 1.14). Dose-dependent increases in AUC0–168h and Cmax occurred in both populations. Accumulation was as expected, based on the half-life (t1/2, ~ 1 week) and dosing interval of semaglutide. Significant body weight reductions were observed with semaglutide 0.5 mg and 1.0 mg in Japanese (both p ≤ 0.05) and Caucasian (both p ≤ 0.05) subjects versus placebo. No new safety issues were identified.
Conclusions
The pharmacokinetic, pharmacodynamic, and safety profiles of semaglutide were similar in Japanese and Caucasian subjects, suggesting that no dose adjustment is required for the clinical use of semaglutide in Japanese subjects.
Funding
Novo Nordisk A/S, Denmark.
Trial registration
ClinicalTrials.gov identifier NCT02146079. Japanese trial registration number JapicCTI-142550.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The prevalence of type 2 diabetes (T2D) and prediabetes has increased significantly since the 1980s in the general Japanese population. The rising incidence of obesity and a marked decline in physical activity have both exerted an influence on this increasing T2D trend [1]. Despite the availability of numerous glucose-lowering drugs, there remains an unmet need for effective diabetes treatment [2], as a significant proportion of individuals with T2D are still not achieving recommended glycemic targets (e.g., HbA1c < 7.0% [American Diabetes Association (ADA) and Japanese Diabetes Society] and ≤ 6.5% [American Association of Clinical Endocrinologists]) [3,4,5].
Glucagon-like peptide (GLP-1) is an incretin hormone that has a glucose-dependent stimulatory effect on insulin and an inhibitory effect on glucagon secretion from the pancreatic islets [6, 7]. The effects of GLP-1 are reduced in individuals with T2D [8,9,10,11]; however, the ability of GLP-1 to lower blood glucose levels is preserved when it is administered at supraphysiologic levels [12]. GLP-1 also has effects on appetite and energy intake [13, 14] and is, therefore, an attractive pharmacologic option for the treatment of T2D [15,16,17]. The short terminal elimination half-life (t1/2) of endogenous GLP-1 (< 1.5 min after intravenous administration [18]) has required the development of GLP-1 receptor agonists with longer half-lives.
GLP-1 receptor agonists have become recognized treatment options for T2D [19], which stimulate insulin and decrease glucagon secretion in a glucose-dependent manner, thereby improving glycemic control with a relatively low risk of hypoglycemia [20].
Semaglutide (Novo Nordisk A/S, Denmark) is a subcutaneously administered GLP-1 analogue for the treatment of T2D. It has 94% amino acid sequence homology to native GLP-1, and is structurally similar to liraglutide, a once-daily GLP-1 receptor agonist [21]. Semaglutide’s structural modifications from native GLP-1 make it less susceptible to degradation by dipeptidyl peptidase 4 (DPP-4) and, in addition, increase its specific high-affinity binding to albumin [21]. The resultant half-life of approximately 1 week makes semaglutide appropriate for once-weekly administration [22, 23].
The phase 3 global SUSTAIN trials were designed to cover a broad spectrum of the T2D patient care continuum, including drug-naïve patients as well as those on a background of orally administered antidiabetic drugs and/or insulin. In these trials, semaglutide was superior to all comparators in reducing HbA1c levels, providing clinically meaningful improvements in glycemic control and body weight reduction in subjects with T2D [24,25,26,27,28]. In SUSTAIN 6—a 2-year placebo-controlled trial investigating cardiovascular outcomes—semaglutide led to a significant 26.0% reduction in risk of the primary outcome (a composite of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke) versus placebo [29].
Clinical pharmacology studies have shown that pharmacokinetic properties of semaglutide are not significantly affected by renal function [30] or hepatic impairment [31], suggesting that no dose adjustment is warranted. Similarly, no clinically relevant impact of semaglutide on concomitant orally administered medications was observed [22, 32].
Genetic differences, diet, and other environmental factors can contribute to differences in therapeutic drug response across different regional populations [33]. Therefore, the objectives of this clinical pharmacology trial were to compare the pharmacokinetics, pharmacodynamics, safety, and tolerability of semaglutide in healthy Japanese and Caucasian subjects using the same doses (0.5 and 1.0 mg) and dose-escalation regimen to reach steady state as in the semaglutide global phase 3 program.
Methods
Trial Design, Subjects, and Dosing
This was a clinical pharmacology, single-center, parallel-group, randomized, double-blind, placebo-controlled trial with two doses of semaglutide (0.5 mg and 1.0 mg) administered subcutaneously to healthy male Caucasian and Japanese subjects.
Healthy male subjects, age 20–55 years, with good glycemic control [HbA1c 6.0% (42 mmol/mol) or less]; body mass index (BMI) in the range of 20–25 kg/m2 with body weight of at least 54 kg; and with both parents either Japanese (for Japanese subjects) or Caucasian (for Caucasian subjects) were eligible for inclusion. Exclusion criteria included the presence or history of endocrine disorders and the existence of a first-degree relative with T2D, and were otherwise standard criteria for clinical pharmacology studies in healthy subjects.
Subjects were randomized within each ethnic group (Japanese and Caucasian) to receive either semaglutide 0.5 mg, semaglutide 1.0 mg, placebo 0.5 mg, or placebo 1.0 mg in an 8:8:3:3 ratio. Subjects were assigned to the lowest available randomization number on a subject-specific prepacked trial product (Clinical Supplies Coordination, Novo Nordisk A/S). Randomization to either active treatment or placebo was double-blinded, but assignment across doses was not blinded, because of differences in treatment volume.
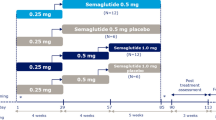
Subjects received once-weekly subcutaneously administered semaglutide or volume-matched placebo for 13 weeks, with 5 weeks’ follow-up (Fig. 1). Doses were administered subcutaneously by the investigator at the clinic, using a prefilled PDS290 pen-injector. Semaglutide steady state was reached using the same dose-escalation regimen as in the phase 3 semaglutide program: subjects assigned to 0.5 mg treatment received 0.25 mg for 4 weeks, then 0.5 mg for 9 weeks. Those in the semaglutide 1.0 mg arm received 0.25 mg for 4 weeks, followed by 0.5 mg for 4 weeks and 1.0 mg for 5 weeks.

Study design. BMI body mass index, PD pharmacodynamic, PK pharmacokinetic, SD single dose, SS steady state
Prior to initiation, the trial protocol and any amendments, the consent form, and the subject information sheet were reviewed and approved according to local regulations by appropriate health authorities, and by an institutional review board (IRB). The IRB for this study was Medical Co. LTA Hakata Clinic IRB, Japan. The trial was conducted in accordance with the Declaration of Helsinki [34] and the International Conference on Harmonization Good Clinical Practice guidelines [35]. Informed consent was obtained before any trial-related activities were initiated.
Trial Endpoints
The primary endpoint of the trial was area under the plasma semaglutide concentration–time curve during a dosing interval (0–168 h) at steady state (AUC0–168h,sema,SS). Secondary pharmacokinetic endpoints for semaglutide at steady state after the last dose of semaglutide, 0.5 mg or 1.0 mg, included maximum plasma concentration (Cmax,sema,SS), time to Cmax,sema,SS (tmax,sema,SS), total apparent clearance (CL/Fsema,SS), terminal elimination half-life (t1/2,sema,SS), and apparent volume of distribution (Vz/Fsema,SS). Secondary pharmacokinetic endpoints after the first dose of semaglutide (0.25 mg) were AUC0–168h,sema,SD, Cmax,sema,SD, and tmax,sema,SD. Other secondary pharmacokinetic endpoints included trough plasma concentration, 168 h after each dose of semaglutide 0.25 mg, 0.5 mg, and 1.0 mg (Ctrough,sema). The dose-corrected accumulation ratio (Racc,DC,sema) was calculated on the basis of AUC0–168h,sema,SD after the first dose and AUC0–168h,sema,SS after the last dose.
The pharmacodynamic endpoints were change from baseline to end of treatment—defined as 1 week after the last dose—in body weight, fasting plasma glucose (FPG), and other fasting glucose metabolism parameters (insulin, C-peptide, glucagon, and pro-insulin). Safety endpoints included incidence of adverse events, number of hypoglycemic episodes, incidence of anti-semaglutide antibodies at follow-up, and change in vital signs, electrocardiogram (ECG) readings, physical examination, and laboratory tests (hematology, biochemistry, calcitonin, and urinalysis).
Pharmacokinetic Assessments
Blood samples for pharmacokinetic assessment of semaglutide were taken before the first dose (0 h), for 1 week following a single 0.25 mg dose (6, 12, 18, 24, 30, 36, 42, 48, 60, 72, 84, 96, 120, 144, 168 h), then weekly prior to the next dose (trough values), and for 5 weeks following the final dose (0 h then 6, 12, 18, 24, 30, 36, 42, 48, 60, 72, 84, 96, 120, 144, 168, 336, 504, 672 and 840 h).
Bioanalysis of blood samples was performed at a specialized laboratory (Celerion Switzerland AG; Fehraltorf, Switzerland), which used a validated liquid chromatography–mass spectrometry (LC–MS/MS) assay [22]. This assay measured the total semaglutide concentration and had a lower limit of quantification (LLOQ) of 0.7 nmol/L [22].
Pharmacodynamic Assessments
Body weight was measured at baseline (before first dosing) and at 8 and 12 weeks after first dosing and 1 week following the final dose. Subjects were weighed without shoes and wearing light clothing. Blood samples for assessment of glucose metabolism parameters (glucose, insulin, C-peptide, glucagon, and pro-insulin) were collected during fasting state: at baseline (before first dosing), 1 week after the fourth dose of 0.5 mg, and 1 week following the final dose. These parameters were analyzed using standardized procedures at a central laboratory (ICON Laboratory Services Inc., Dublin, Ireland) while Celerion Switzerland AG was responsible for the analysis of semaglutide and anti-semaglutide antibodies. Local laboratory facilities (Medical Co. LTA, Sumida Hospital; LSI Medscience Cooporation, and SRL Inc. [all Tokyo, Japan]) provided additional analyses including biochemistry, hematology, urinalysis, and other laboratory assessments.
Safety Assessments
Safety assessments included monitoring of adverse events/serious adverse event and vital signs, ECG, physical examination, hypoglycemic episodes, and laboratory testing (including presence of anti-semaglutide antibodies or hypersensitivity). All adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA), version 17.1. Hypoglycemia was monitored at the trial site during the in-house visits by glucose measurement. Hypoglycemia was classified as severe according to ADA guidelines [36] or blood glucose-confirmed symptomatic hypoglycemia by a plasma glucose value of less than 56 mg/dL (3.1 mmol/L) with symptoms indicative of hypoglycemia.
Statistical Analysis
Sample size was not based on a formal statistical power calculation. A total of 44 subjects (22 Japanese and 22 Caucasian) were planned to be randomized to semaglutide 1.0 mg (n = 8), semaglutide 0.5 mg (n = 8), placebo 0.5 mg (n = 3), or placebo 1.0 mg (n = 3). In the analysis, the two placebo groups were pooled assuming no correlation between endpoints and placebo volume. A withdrawal rate of 15.0% was anticipated, which allowed for at least six steady-state pharmacokinetics profiles per dose and race group to be available for evaluation. All subjects who were randomized and exposed to at least one dose of trial product contributed to the pharmacokinetics, pharmacodynamics, and safety analyses.
The pharmacokinetic primary endpoint, AUC0–168h,sema,SS, was derived from the concentration–time curve during a dosing interval (0–168 h) at steady state using the linear trapezoidal method on the observed concentrations at actual time points. The AUC0–168h,sema,SS and other steady-state pharmacokinetic endpoints (Cmax,sema,SS and Racc,DC,sema) were analyzed separately by linear normal models based on log-transformed values with race (Japanese or Caucasian), dose group (0.5 mg or 1.0 mg), and race-by-dose group interaction as fixed factors. Estimated race differences (Japanese/Caucasian) by dose group and estimated dose-group differences (1.0 mg/0.5 mg) by race were transformed back to the original scale and presented as ratios with two-sided 95% confidence intervals (CI). A p value for the test of race-by-dose group interaction was also calculated. The race ratio for the pharmacokinetic endpoints (AUC0–168h,sema,SD and Cmax,sema,SD) after the first dose (0.25 mg) was estimated by linear normal models on log-transformed values.
For each pharmacodynamic assessment, treatment contrasts (semaglutide 0.5 mg and 1.0 mg versus placebo) at the end of treatment were estimated using a mixed model for repeated measurements using all post-baseline measurements up to the end of treatment. The model included visit, dose group, race, and race-by-dose interaction as fixed factors and baseline value as covariate. Furthermore, interaction terms of visit-by-dose group, visit-by-race, visit-by-race-by-dose group, and visit-by-baseline value were included. Fasting insulin, C-peptide, glucagon, and pro-insulin were logarithmically transformed.
Safety endpoints were summarized descriptively. We used SAS version 9.3 for all analyses.
Results
Subject Disposition and Baseline Characteristics
In total, 44 subjects (Japanese, n = 22; Caucasian, n = 22) were randomized and exposed to the trial product between May and October 2014, from 108 subjects initially screened for eligibility. Three Caucasian subjects withdrew from the trial for personal reasons: one in the semaglutide 0.5 mg group (day 50) and two in the semaglutide 1.0 mg group (days 29 and 36). Baseline characteristics and demographics are shown in Table 1. Caucasian subjects were on average taller and weighed more than their Japanese counterparts (73.0 versus 63.6 kg, respectively). There were no other marked differences in baseline characteristics between dose groups, and BMI levels were comparable between the populations. In line with the criteria for healthy volunteers, glycemic parameters were within the normal range. Baseline insulin, pro-insulin, C-peptide, and glucagon levels were also similar between race groups.
Pharmacokinetics
Plasma levels of semaglutide were detectable in all semaglutide-treated subjects, and below the assay LLOQ in all subjects receiving placebo. The full plasma concentration–time profile of semaglutide, from first dose until follow-up, is shown in Fig. 2. Mean semaglutide concentration profile was comparable between race groups throughout the trial period at both dose levels. Semaglutide concentrations increased from day 28, reflecting the dose-escalation step from 0.25 to 0.5 mg in all groups. Similarly, mean semaglutide concentrations increased from day 56 in the 1.0 mg groups, following the final dose-escalation step from 0.5 to 1.0 mg. Pharmacokinetic samples were taken more frequently for 0–168 h following the first dose (day 0) and 0–840 h after the last dose (day 84), as indicated by the graph peaks (Fig. 2).

Mean semaglutide profile from first dosing to follow-up. Values are geometric means. From day 7 to day 84 all samples were taken immediately prior to next semaglutide dose (trough samples). All values below the lower limit of quantification are imputed
The mean semaglutide concentration profile during the dosing interval at steady state, from day 84, was similar in Japanese and Caucasian subjects within each dose group (Fig. 3). The exposure of semaglutide at steady state (AUC0–168h,sema,SS) was comparable between Japanese and Caucasian subjects in both dose groups (Table 2), with similar estimated race ratios for semaglutide 0.5 and 1.0 mg doses (Table 3). There was a dose-dependent increase for AUC0–168h,sema,SS in both populations, which is in accordance with dose proportionality (Table 2), as shown by estimated treatment ratios (semaglutide 1.0 mg/semaglutide 0.5 mg) that were close to 2 for both populations (Table 3). There was no statistically significant race-by-dose group interaction.

Mean semaglutide profile at steady state: 0–168 h after last dose. Values are geometric means
The Cmax,sema,SS of semaglutide at steady state was also similar between Japanese and Caucasian subjects (Table 2), with estimated race ratios close to 1 and estimated treatment ratios close to 2 (Table 3). Other pharmacokinetic endpoints at steady state (tmax,sema,SS, CL/Fsema,SS) were comparable between the populations in both dose groups (Table 2). The elimination of semaglutide during follow-up, following the final dose, is shown in Supplementary Fig. S1. Although the Vz/Fsema,SS of semaglutide was similar between the populations and doses, the half-life of semaglutide (t1/2,sema,SS) appeared to be slightly lower in the 0.5 mg Japanese group versus the other dose groups (Table 2).
The exposure (AUC0–168h,sema,SD) and Cmax,sema,SD after a single dose (0.25 mg) of semaglutide was slightly higher in Japanese versus Caucasian subjects, as shown by estimated race ratios above 1 (Supplementary Table S1; Supplementary Fig. S2) and the tmax,sema,SD was earlier for Japanese subjects than for Caucasian subjects.
Trough plasma concentrations (Ctrough,sema concentration 1 week after the fourth dose of all dose levels) were similar between race groups and across doses, and trough levels were greater with the higher 1.0 mg dose upon dose escalation (Fig. 2). The dose-corrected accumulation ratio (Racc, DC,sema) was slightly lower in Japanese subjects than in Caucasian subjects (Table 3) but was comparable among dose groups (0.5 and 1.0 mg).
Pharmacodynamics
Mean body weight was decreased by 1.4 and 5.0 kg with semaglutide 0.5 and 1.0 mg, respectively, compared with an increase of 1.1 kg with placebo in Japanese subjects [estimated treatment difference versus placebo − 2.4 kg (p ≤ 0.05) and − 6.1 kg (p ≤ 0.05)]. In Caucasian subjects, mean body weight was reduced by 3.6 kg with semaglutide 0.5 mg and 7.5 kg with semaglutide 1.0 mg, versus an increase of 0.7 kg with placebo [estimated treatment difference − 4.3 kg (p ≤ 0.05) and − 8.3 kg (p ≤ 0.05)] (Supplementary Table S2).
No clinically significant changes in the different parameters of glucose metabolism from baseline to end of treatment were observed. There was a slight reduction in FPG in Japanese subjects receiving semaglutide 0.5 and 1.0 mg, compared with placebo and in Caucasian subjects receiving semaglutide 1.0 mg (Supplementary Table S2).
Safety
Overall, 32 subjects were exposed to semaglutide and 12 subjects to placebo across both populations. No deaths, serious adverse events, or adverse events leading to withdrawal were reported. The proportions of Japanese subjects reporting adverse events were 75.0% and 62.5% with semaglutide 0.5 and 1.0 mg, respectively, compared with 16.7% of subjects receiving placebo. In the Caucasian population, 50.0%, 62.5%, and 33.3% of subjects reported adverse events with semaglutide 0.5, 1.0 mg, and placebo, respectively (Supplementary Table S3). The number of events was higher in the semaglutide 1.0 mg group, compared with the 0.5 mg group (driven by three subjects having multiple events), with no apparent difference between the race groups. All adverse events were mild to moderate in severity; none were severe. Gastrointestinal adverse events were the most frequently reported events, and more common with semaglutide than placebo. A higher proportion of Japanese subjects reported gastrointestinal adverse events than Caucasian subjects: 62.5%, 37.5%, and 16.7% of Japanese subjects receiving semaglutide 0.5, 1.0 mg and placebo, respectively, versus 25.0%, 25.0% and 0.0% of Caucasian subjects, respectively (Supplementary Table S3). No hypoglycemic events were reported during the trial.
There was an elevation in pulse rate from baseline to end of treatment in subjects receiving semaglutide, with no apparent difference across the races and doses. Pulse rate increased by 8.5 beats per minute (bpm) and 10.4 bpm in Japanese subjects receiving semaglutide 0.5 and 1.0 mg, respectively, versus a decrease of 0.5 bpm with placebo. In Caucasian subjects, pulse rate increased by 14.6, 10.5, and 2.8 bpm with semaglutide 0.5, 1.0 mg, and placebo, respectively.
There were no clinically significant abnormalities in ECG readings or physical examinations. No noticeable trends were found in the biochemistry parameters, including amylase, lipase and calcitonin, in any of the treatment or race groups. No subjects developed anti-semaglutide antibodies as a consequence of semaglutide treatment.
Discussion
This trial investigated whether the pharmacokinetics, pharmacodynamics, safety, and tolerability profile of semaglutide in healthy male Japanese subjects were similar to those in Caucasian subjects. The primary objective was to assess and compare exposure after multiple doses of semaglutide using the clinical therapeutic doses of 0.5 and 1.0 mg.
Semaglutide pharmacokinetic profiles and pharmacokinetic parameters were similar across both populations, and showed an expected higher exposure with the 1.0 mg dose, as demonstrated by the mean semaglutide concentration–time curves from first dosing to follow-up, which is supported by the statistical analyses. Furthermore, the pharmacokinetic properties of semaglutide reported in this clinical trial were comparable with those reported from other reported clinical pharmacology trials [22, 30,31,32].
Following multiple doses of semaglutide, the Cmax and tmax were found to be comparable between the races, despite a slightly higher mean semaglutide exposure and Cmax after the first single 0.25 mg semaglutide dose in Japanese subjects.
The dose-corrected accumulation ratio of approximately 2 was as expected on the basis of the half-life of semaglutide of approximately 1 week [21] and the once-weekly dosing frequency. Semaglutide 1.0 mg resulted in double the exposure of the 0.5 mg dose, with no difference between the two races.
Dose-dependent reductions in body weight were observed in all subjects receiving active treatment; however, this was more pronounced in Caucasian subjects, who had a higher mean body weight at baseline. Weight reduction is a known effect of GLP-1 receptor agonist therapies, and some, such as liraglutide, are licensed to treat individuals with obesity in some countries [37, 38]. Semaglutide was shown to provide significant and clinically meaningful improvements in glycemic control and body weight reduction in the global phase 3 trials versus all comparators [24,25,26,27,28], and in phase 3 trials of Japanese subjects with T2D versus sitagliptin and orally administered antidiabetic agent add-on therapy [39, 40]. As would be expected in a trial population of healthy volunteers, there were no clinically significant changes in glucose metabolism in the current trial.
No new safety and tolerability issues related to semaglutide were observed in this trial. There were no serious adverse events and no subjects withdrew because of adverse events, while three withdrew for personal reasons. Although semaglutide-treated Japanese subjects reported more gastrointestinal adverse events, the overall safety and tolerability profile of semaglutide was comparable between Japanese and Caucasian subjects and was similar to that reported in other semaglutide trials [24,25,26,27,28]. In line with results from other trials, gastrointestinal adverse events—an established side effect of GLP-1 receptor agonist therapy [41]—were reported more frequently with semaglutide.
In this trial of healthy volunteers, there was an elevation in pulse rate in semaglutide-treated subjects; however, considering the low sample size, the change observed in Japanese and Caucasian subjects was comparable. Only minor increases in pulse rate from baseline were observed across the phase 3 SUSTAIN program in subjects with T2D [24,25,26,27,28]. In SUSTAIN 6, the largest trial investigating semaglutide, with the longest duration reported to date in subjects with high cardiovascular risk, mean pulse rate increases of 2.1 and 2.4 bpm were reported with semaglutide 0.5 and 1.0 mg, respectively, versus no change with placebo. Despite this slight elevation, it should be noted that semaglutide-treated patients had a 26.0% lower risk of the primary composite outcome (death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke) than those who received placebo [29].
GLP-1 receptor agonists are known to affect pulse rate [42] and, accordingly, when semaglutide was compared head-to-head with another once-weekly GLP-1 receptor agonist, exenatide extended release, the elevation in pulse rate was not significantly different [26]. There were no cases of positive anti-semaglutide antibodies or episodes of hypoglycemia reported during this trial.
This trial had a relatively short duration and a small sample size. Furthermore, the subjects enrolled were healthy, lean individuals with no expected effects on pharmacodynamic parameters. A key limitation of the study is that participants were not entirely representative of the population for which semaglutide treatment is intended, in individuals with T2D. It has been proposed that the pathophysiology of T2D in Japanese patients is primarily characterized by β-cell dysfunction, while obesity and insulin resistance are believed to be the key drivers of T2D in Caucasians [43]. As semaglutide improves β-cell function and corrects insulin secretion [44], these actions may be highly beneficial to Japanese patients [45]. The efficacy and safety of semaglutide in Japanese patients with T2D have previously been reported in larger, phase 3 trials [39, 40]. These investigations showed that the responses were similar in the two populations. It may be of clinical interest to understand if there are any differences in mechanism of action across these different populations in regards to improving glycemic control.
In summary, while this study reached its objective and enabled the analysis of pharmacokinetics and safety across healthy Japanese and Caucasian subjects, future studies may investigate the differences in pharmacokinetics and pharmacodynamics across Japanese and Caucasian patients with T2D.
Conclusion
Following multiple doses of semaglutide 0.5 and 1.0 mg, exposure of semaglutide at steady state was comparable between Japanese and Caucasian subjects, with an expected dose-dependent increase. The safety findings were comparable across the populations. These findings suggest that no dose adjustment would be required for the clinical use of semaglutide in Japanese subjects.
References
Mukai N, Ninomiya T, Hirakawa Y, et al. Trends in the prevalence of type 2 diabetes and prediabetes in community-dwelling Japanese subjects: the Hisayama study. J Diabetes Invest. 2014;5:162–9.
Stark Casagrande S, Fradkin JE, Saydah SH, Rust KF, Cowie CC. The prevalence of meeting A1C, blood pressure, and LDL goals among people with diabetes, 1988–2010. Diabetes Care. 2013;36:2271–9.
American Diabetes Association. Glycemic targets. Sec. 5. In standards of medical care in diabetes. Diabetes Care. 2016;39(Supplement 1):S39–46.
The Japan Diabetes Society. Treatment guide for diabetes 2014–2015: Bunkodo. http://www.jds.or.jp/modules/en/index.php?content_id=1. Accessed Feb 2018.
Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm-2016 executive summary. Endocr Pract. 2016;22:84–113.
Holst JJ, Vilsbøll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol. 2009;297:127–36.
Kieffer TJ, Francis Habener J. The glucagon-like peptides. Endocr Rev. 1999;20:876–913.
Bagger JI, Knop FK, Lund A, Vestergaard H, Holst JJ, Vilsbøll T. Impaired regulation of the incretin effect in patients with type 2 diabetes. J Clin Endocrinol Metab. 2011;96:737–45.
Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29:46–52.
Nauck M, Vardarli I, Deacon C, Holst JJ, Meier J. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: what is up, what is down? Diabetologia. 2011;54:10–8.
Perley MJ, Kipnis DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic subjects. J Clin Investig. 1967;46:1954.
Højberg P, Vilsbøll T, Rabøl R, et al. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia. 2009;52:199–207.
Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Investig. 1998;101:515.
Bagger JI, Holst JJ, Hartmann B, Andersen B, Knop FK, Vilsboll T. Effect of oxyntomodulin, glucagon, GLP-1, and combined glucagon +GLP-1 infusion on food intake, appetite, and resting energy expenditure. J Clin Endocrinol Metab. 2015;100:4541–52.
Nauck M, Kleine N, Ørskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1993;36:741–4.
Nauck MA. Incretin-based therapies for type 2 diabetes mellitus: properties, functions, and clinical implications. Am J Med. 2011;124:S3–18.
Toft-Nielsen M-B, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab. 2001;86:3717–23.
Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87:1409–39.
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140–9.
Meloni AR, DeYoung MB, Lowe C, Parkes DG. GLP-1 receptor activated insulin secretion from pancreatic β-cells: mechanism and glucose dependence. Diabetes Obes Metab. 2013;15:15–27.
Lau J, Bloch P, Schaffer L, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58:7370–80.
Kapitza C, Nosek L, Jensen L, Hartvig H, Jensen CB, Flint A. Semaglutide, a once-weekly human GLP-1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J Clin Pharmacol. 2015;55:497–504.
Kapitza C, Dahl K, Jacobsen J, Axelsen M, Flint A. Effects of semaglutide on beta-cell function and glycaemic control in participants with type 2 diabetes: a randomised, double-blind, placebo-controlled trial. Diabetologia. 2017;60:1390–9.
Sorli C, Harashima SI, Tsoukas GM, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5:251–60.
Ahrén B, Comas LM, Kumar H, et al. Efficacy and safety of once-weekly semaglutide versus once-daily sitagliptin as an add-on to metformin, thiazolidinediones, or both, in patients with type 2 diabetes (SUSTAIN 2): a 56-week, double-blind, phase 3a, randomised trial. Lancet Diabetes Endocrinol. 2017;5:341–54.
Ahmann A, Capehorn M, Charpentier G, et al. Efficacy and safety of once-weekly semaglutide versus exenatide ER in subjects with type 2 diabetes (SUSTAIN 3): a 56-week, open-label, randomized clinical trial. Diabetes Care. 2018;41:258–66.
Aroda V, Bain S, Cariou B, et al. Efficacy and safety of once-weekly semaglutide versus once-daily insulin glargine as add-on to metformin (with or without sulfonylureas) in insulin-naive patients with type 2 diabetes (SUSTAIN 4): a randomised, open-label, parallel-group, multicentre, multinational, phase 3a trial. Lancet Diabetes Endocrinol. 2017;5:355–66.
Rodbard H, Lingvay I, Reed J, et al. Efficacy and safety of semaglutide once-weekly vs placebo as add-on to basal insulin alone or in combination with metformin in subjects with type 2 diabetes (SUSTAIN 5). Diabetologia. 2016;59(Suppl 1):S364–5.
Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;15(10):1834–44.
Marbury T, Flint A, Segel S, Lindegaard M, Lasseter K. Pharmacokinetics and tolerability of a single dose of semaglutide, a human glucagon-like peptide-1 analog, in subjects with and without renal impairment. Clin Pharmacokinet. 2017. https://doi.org/10.1007/s40262-017-0528-2.
Jensen L, Kupčová V, Arold G, Pettersson J, Hjerpsted J. Pharmacokinetics and tolerability of semaglutide in subjects with hepatic impairment. Diabetes Obes Metab. 2017. https://doi.org/10.1111/dom.13186.
Hausner H, Derving Karsbøl J, Anderson TW, et al. Effect of once-weekly subcutaneous treatment with semaglutide, a GLP-1 analog, on metformin, warfarin, atorvastatin and digoxin in healthy subjects. In: ENDO 2016—98th Annual Meeting of the Endocrine Society. 2016;37(3):FR-710.
Yasuda SU, Zhang L, Huang SM. The role of ethnicity in variability in response to drugs: focus on clinical pharmacology studies. Clin Pharm Ther. 2008;84:417–23.
World Medical Association. Declaration of Helsinki ethical principles for medical research involving human subjects 52nd WMA general assembly, Edinburgh, Scotland, October 2000. Last amended with Note of Clarification on Paragraph 29 by the WMA General Assembly, Washington 2002; and Note of Clarification on Paragraph 30 by the WMA General assembly, Tokyo 2004. https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/. Accessed Feb 2018.
International Conference on Harmonisation Working Group. ICH harmonised tripartite guideline: guideline for good clinical practice E6 (R1). 1996. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E_R1_Guideline.pdf. Accessed Oct 2016.
American Diabetes Association. Standards of medical care in diabetes. Diabetes Care. 2017;40(Suppl 1):S1–135.
Novo Nordisk. Saxenda: liraglutide injection 3 mg 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206321Orig1s000lbl.pdf. Accessed Feb 2018.
Pi-Sunyer X, Astrup A, Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373:11–22.
Seino Y, Terauchi Y, Osonoi T, et al. Safety and efficacy of semaglutide once weekly vs sitagliptin once daily, both as monotherapy in Japanese people with type 2 diabetes. Diabetes Obes Metab. 2018;20:378–88.
Kaku K, Yamada Y, Watada H, et al. Safety and efficacy of once-weekly semaglutide versus additional oral antidiabetic drugs, in Japanese subjects with inadequately controlled T2D: a randomised trial. Diabetes Obes Metab. 2018. https://doi.org/10.1111/dom.13218.
Horowitz M, Aroda VR, Han J, Hardy E, Rayner CK. Upper and/or lower gastrointestinal adverse events with GLP-1 receptor agonists: incidences and consequences. Diabetes Obes Metab. 2017;19:672–81.
Robinson LE, Holt TA, Rees K, Randeva HS, O’Hare JP. Effects of exenatide and liraglutide on heart rate, blood pressure and body weight: systematic review and meta-analysis. BMJ Open. 2013. https://doi.org/10.1136/bmjopen-2012-001986.
Yabe D, Seino Y, Fukushima M, Seino S. Beta cell dysfunction versus insulin resistance in the pathogenesis of type 2 diabetes in East Asians. Curr Diab Rep. 2015;15:602.
Kapitza C, Dahl K, Jacobsen JB, Axelsen MB, Flint A. Effects of semaglutide on beta cell function and glycaemic control in participants with type 2 diabetes: a randomised, double-blind, placebo-controlled trial. Diabetologia. 2017;60:1390–9.
Seino Y, Kuwata H, Yabe D. Incretin-based drugs for type 2 diabetes: focus on East Asian perspectives. J Diabetes Investig. 2016;7(Suppl 1):102–9.
Acknowledgements
Funding
Sponsorship for this study, article processing charges, and the open access fee were funded by Novo Nordisk, A/S.
Editorial Assistance
We also thank Gurudutt Nayak (Novo Nordisk A/S, Denmark) for his review and input to the manuscript, and Nicole Antonio, PhD (AXON Communications, UK) for medical writing and editorial assistance. Support for this assistance was funded by Novo Nordisk.
Authorship
All of the named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published. All authors had full access to all of the data in this study and take full responsibility for the integrity of the data and accuracy of the data analysis.
Thanks
We thank all the participants, investigators, and trial-site staff from Medical Co. LTA, Sumida Hospital in Tokyo, Japan who were involved in the conduct of the trial.
Disclosures
Shin Irie’s institution has received funding to conduct clinical trials from Novo Nordisk, Eli Lilly and Company, Teijin Pharma, Daiichi Sankyo Company, Takeda Pharmaceutical Company Limited, Sanofi K.K., Nippon Boehringer Ingelheim Co., and AstraZeneca. Ippei Ikushima’s institution has also received funding to conduct clinical trials from Novo Nordisk, Eli Lilly and Company, Teijin Pharma, Daiichi Sankyo Company, Takeda Pharmaceutical Company Limited, Sanofi K.K., Nippon Boehringer Ingelheim Co., and AstraZeneca. Lene Jensen is an employee and shareholder of Novo Nordisk A/S. Anne Flint is an employee and shareholder of Novo Nordisk A/S. Jeppe Zacho is an employee and shareholder of Novo Nordisk A/S. Tomoyuki Nishida is an employee of Novo Nordisk Pharma Ltd. and shareholder of Novo Nordisk A/S.
Compliance with Ethics Guidelines
Prior to initiation, the trial protocol and any amendments, the consent form, and the subject information sheet were reviewed and approved according to local regulations by appropriate health authorities, and by an institutional review board (IRB). The IRB for this study was Medical Co. LTA Hakata Clinic IRB, Japan.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments and the International Conference on Harmonization Good Clinical Practice guidelines. Informed consent was obtained from all individual participants included in the study.
Data Availability
The datasets generated and/or analyzed during the current study are not publicly available as this was a phase I study, but are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to https://doi.org/10.6084/m9.figshare.5882737.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ikushima, I., Jensen, L., Flint, A. et al. A Randomized Trial Investigating the Pharmacokinetics, Pharmacodynamics, and Safety of Subcutaneous Semaglutide Once-Weekly in Healthy Male Japanese and Caucasian Subjects. Adv Ther 35, 531–544 (2018). https://doi.org/10.1007/s12325-018-0677-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-018-0677-1