
Abstract
Introduction
Mifepristone, a competitive glucocorticoid receptor antagonist approved for Cushing syndrome, and ketoconazole, an antifungal and steroidogenesis inhibitor, are both inhibitors of and substrates for cytochrome P450 (CYP3A4). This study evaluated the pharmacokinetic effects of concomitant ketoconazole, a strong CYP3A4 inhibitor, on mifepristone.
Methods
In an open-label, two-period, single-center study, healthy adult men received mifepristone 600 mg orally daily for 12 days (period 1) followed by mifepristone 600 mg daily plus ketoconazole 200 mg orally twice daily for 5 days (period 2). Serial pharmacokinetic blood samples were collected predose and over 24 h postdose on days 12 (period 1) and 17 (period 2). A cross-study comparison (using data on file) further examined whether systemic exposure to mifepristone plus ketoconazole exceeded the exposure following mifepristone 1200 mg orally administered for 7 days.
Results
Sixteen subjects were enrolled and 14 completed the study. Concomitant administration with ketoconazole increased the systemic exposure to mifepristone, based on geometric least squares mean ratios, by 28% for C max and 38% for AUC0–24. This increase was 85% and 87% of the exposure observed following mifepristone’s highest label dose of 1200 mg/day for C max and AUC0–24, respectively. Adverse events (AEs) were reported in 56.3% (9/16) of subjects during administration of mifepristone alone and in 57.1% (8/14) during combination with ketoconazole. No serious AEs were reported.
Conclusion
Systemic exposure to mifepristone increased following multiple doses of mifepristone 600 mg daily plus ketoconazole 200 mg twice daily. Little to no increase in AEs occurred. Dose adjustment of mifepristone may be needed when given with ketoconazole.
Funding
Corcept Therapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mifepristone (Korlym®, Menlo Park, California) is a competitive glucocorticoid receptor antagonist that has demonstrated clinical efficacy and safety in patients with Cushing syndrome (CS) and diabetes or glucose intolerance and who cannot undergo surgery or have failed surgery [1]. The pharmacokinetic profile of mifepristone is characterized as nonlinear with less than dose-proportional increases in serum concentration following increased single or multiple doses [2, 3]. Following oral administration, mifepristone is rapidly absorbed with time to peak plasma concentrations between 1 and 2 h after single doses and 1 and 4 h after multiple doses [2, 4]. The half-life of mifepristone is ~ 85 h, with time to steady state achieved within 2 weeks [4].
Mifepristone is metabolized primarily via cytochrome P450 (CYP) isoform 3A4, yielding three active metabolites RU 42633 (mono-demethylated), RU 42848 (di-demethylated), and RU 42698 (hydroxylated) (Fig. 1) [5, 6]. The metabolites are each less potent than mifepristone at the glucocorticoid receptor; the affinities of RU 42633, RU 42848, and RU 42698 to the glucocorticoid receptor are 61%, 45%, and 48%, respectively, compared with 100% for mifepristone [7]. Mifepristone has also been shown under in vitro conditions to inhibit as well as induce CYP3A4 [8, 9]. In addition to CS, mifepristone has also been studied in other patient populations, including those with psychotic depression [10,11,12], and it is currently being evaluated as a concomitant treatment in certain forms of breast and prostate cancers (ClinicalTrials.gov NCT02014337, NCT02788981, NCT02012296) [13,14,15]. Because mifepristone is a substrate of CYP3A4, it is important to examine the effects of a strong CYP3A4 inhibitor on its pharmacokinetic parameters. Typically an area under the curve (AUC) increase of at least 5-fold in the presence of a strong inhibitor indicates a sensitive substrate of CYP3A4 [16]. Data on the pharmacokinetic effects of concomitant administration with a strong CYP3A4 inhibitor on mifepristone are, however, lacking.

Mifepristone and its three major metabolites
Ketoconazole (Nizoral®, Titusville, New Jersey) is a substrate for and well-known strong inhibitor of CYP3A4 [17], and it is listed by the Food and Drug Administration (FDA) as an interacting drug to investigate potential drug–drug interactions between CYP3A4 substrates [16]. Although approved as an antifungal, ketoconazole is sometimes used off-label as a steroidogenesis inhibitor in the treatment of CS [18, 19]. Thus, the assessment of the pharmacokinetic effects of concomitant ketoconazole on mifepristone are of particular interest to clinicians treating patients with CS.
This study, conducted in healthy adult men, evaluated the effects of steady-state ketoconazole on the pharmacokinetics of steady-state mifepristone. The effects of mifepristone on the pharmacokinetics of ketoconazole were also examined. In addition, because mifepristone use is associated with increases in cortisol and adrenocorticotropic hormone (ACTH) levels [1], we examined the effects of concomitant administration of mifepristone and ketoconazole on these parameters in comparison with mifepristone alone. Safety and tolerability of the coadministered agents were also reported.
Methods
Subjects
Eligible participants included healthy men 18–45 years of age with a body mass index (BMI) of 19–32 kg/m2 who weighed at least 60 kg and had no clinically significant abnormal findings on physical examination, electrocardiogram (ECG), blood pressure, heart rate, medical history, or clinical laboratory assessments. Subjects must have had a corrected QT interval no greater than 450 ms on ECG at screening, morning serum cortisol within normal limits, and normal serum potassium. Participants with partners of childbearing potential must have been using two forms of medically accepted methods of contraception throughout the study and for at least 3 months (90 days) after the last dose of study medication; subjects with pregnant partners were excluded. Subjects with any alkaline phosphatase, total bilirubin, or direct bilirubin outside normal range or an alanine aminotransferase or aspartate aminotransferase value greater than 1.2 times the upper limit were also excluded. Additional exclusion criteria included consumption of alcohol or caffeine-containing products 72 h prior to study baseline or use of concomitant medications or nicotine-containing products.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients for being included in the study. The protocol was approved by an independent investigational review board (IntegReview, Austin, Texas). This manuscript was prepared according to the International Society for Medical Publication Professionals’ “Good Publication Practice for Communicating Company-Sponsored Medical Research: the GPP3 Guidelines” and the International Committee of Medical Journal Editors’ “Uniform Requirements for Manuscripts Submitted to Biomedical Journals.”
Study Design
This was a phase 1, open-label, two-period study conducted at a single US center to evaluate the pharmacokinetic effects in healthy men of multiple doses of ketoconazole 200 mg twice daily orally on concomitantly orally administered multiple doses of mifepristone 600 mg once daily and vice versa. A cross-study comparison using data on file was also conducted to determine whether the effect of ketoconazole on the systemic exposure to coadministered mifepristone exceeded that of the maximum recommended dose of mifepristone (1200 mg) [4].
The 600-mg mifepristone dose was selected for analysis based on internal data suggesting that the increased mifepristone exposure resulting from concomitant administration with ketoconazole 400 mg total daily would be generally comparable to the exposure following 1200 mg of mifepristone. The 200-mg twice-daily ketoconazole dose was consistent with the highest clinical dosing recommendations in its product labeling [20]. The twice-daily dosing regimen was also used to enhance the inhibitory effect of ketoconazole when given with a substrate with a long half-life, as is the case for mifepristone [21].
Food can enhance the absorption of mifepristone and ketoconazole [4, 22]; therefore, all medications were administered within 30 min of a typical breakfast consisting of 34% fat. Subjects were admitted to the clinic on day − 2. On day − 1, subjects received a single dose of ketoconazole 200 mg, followed by a 48-h pharmacokinetic sampling period that was completed on day 1 prior to mifepristone administration (Fig. 2). Subjects were discharged from the clinic following the 12-h sample and returned for the 24-, 36-, and 48-h sample. During period 1, mifepristone 600 mg was administered alone on days 1–12. During period 2, which followed on days 13–17, ketoconazole 200 mg twice daily was administered concomitantly with mifepristone 600 mg once daily. The morning dose of ketoconazole was administered approximately 5 min before mifepristone. Subjects were readmitted to the clinic on the evening of day 11 and remained onsite through day 19. Subjects returned to the clinic for pharmacokinetic sampling on days 20, 22, 25, and 28 and a termination visit was conducted on day 32.

Dosing schedule. BID twice daily, QD once daily
Sampling Procedures
Plasma trough concentrations of mifepristone and its three active metabolites were measured from blood samples collected 30 min predose on days 1–11 and days 13–16; on washout days 18, 20, 22, 25, and 28; and on termination visit day 32. Serial blood samples were collected predose and at 0.5, 1, 2, 4, 8, 12, and 24 h on days 12 and 17.
Plasma concentrations of ketoconazole were measured in blood samples collected within 30 min predose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 h on day − 1 and within 30 min predose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 12 h on day 17. Plasma trough concentrations of ketoconazole were also measured 30 min predose from blood samples taken on days 13–16 to ensure steady-state concentrations were reached. In the reference study of mifepristone 1200 mg, plasma concentrations of mifepristone and its metabolites were measured in blood samples collected predose and at 0.5, 1, 2, 4, 8, 12, and 24 h after dosing on days 1 and 7.
Samples were collected in heparinized vacutainer tubes and centrifuged at 2500 rpm for 15 min at 4–6 °C. Harvested plasma samples were stored at approximately − 20 °C or lower and shipped under dry ice for analysis.
Fasting blood samples for the analysis of serum cortisol and plasma ACTH were collected between 07:00 and 09:00 during screening and on days 1 and 6 of period 1, days 13–17 of period 2, and termination day 32. Samples were collected prior to dosing during the study periods.
Bioanalytical Procedures
Plasma concentrations of mifepristone, its metabolites, and ketoconazole were assessed by MicroConstants (San Diego, California) using validated high-performance liquid chromatography (HPLC) methods with mass spectrometric (MS) detection. The assay lower limit of quantification was 10 ng/mL for mifepristone and the three active metabolites, and 5.0 ng/mL for ketoconazole. Data analysis was performed using MassLynx v.4.1 (Waters, Milford, Massachusetts).
Human plasma samples containing C-1073, RU 42633, RU 42698, RU 42848, a solution containing C-1073-d4, RU 42633-d4, RU 42698-d4, and RU 42848-d4 (deCODE Genetics, Reykjavik, Iceland) as the internal standards, and sodium heparin as the anticoagulant were extracted using a mixture of hexane and methyl tert-butyl ether (MTBE). The organic layer was dried down under nitrogen and the residue was reconstituted in water/acetonitrile/formic acid (75:25:0.1, v/v/v). Sample extracts were analyzed by reversed-phase chromatography using a phenyl column maintained at 50 °C. The mobile phase was nebulized using heated nitrogen in a Z-spray source/interface set to electrospray positive ionization mode. The ionized compounds were detected using a tandem quadrupole mass spectrometer (MS/MS).
For determination of ketoconazole concentrations, plasma samples containing ketoconazole-d3 or ketoconazole-d8 (Toronto Research Chemicals Inc., North Yolk, Ontario, Canada) as the internal standard and tripotassium ethylenediaminetetraacetic acid or sodium heparin as the anticoagulant were adjusted to approximately pH 10.0 with ammonium hydroxide and extracted with an MTBE solution. The samples were vortex mixed and centrifuged, and the lower portion was frozen in an ultra-cold freezer. The organic layer was transferred to a clean tube and then evaporated under nitrogen. The residue was reconstituted with acetonitrile. An aliquot was analyzed by reversed-phase HPLC using an Atlantis Hilic Silica column (Waters, Milford, Massachusetts), maintained at 40 °C. The mobile phase was nebulized using heated nitrogen in a Z-spray source/interface and the ionized compounds were detected using MS/MS.
Accuracy and precision (percentage of variation, %CV) were evaluated using replicate analyses of human plasma quality control samples prepared at concentrations of 30.0, 300, and 1600 ng/mL for mifepristone and its metabolites and 15.0, 200, and 4000 ng/mL for ketoconazole. Deviation of the measured concentration from the theoretical values (range ± 2.00% to ± 8.67%) and %CV (range 1.82–6.27%) of quality control samples of mifepristone, its metabolites (RU 42633, RU 42698, and RU 42848), and ketoconazole were all within 15%.
Pharmacokinetic Assessments
For each individual, pharmacokinetic parameters were computed for mifepristone and its metabolites and ketoconazole using a non-compartmental analysis method using WinNonlin version 6.4 (Certara Inc, St. Louis, Missouri). Parameters included maximum plasma concentration (C max), area under the curve (AUC) using the linear trapezoidal rule, and time to maximum plasma concentration (t max). For ketoconazole, AUC was calculated as AUC extrapolated to infinity (AUCinf) for day − 1 and AUC0–12 for day 17.
Safety
Safety and tolerability were assessed throughout the study by physical examinations, clinical laboratory tests, vital signs, ECGs, and adverse event (AE) reporting. The causal relationship of each AE to mifepristone and/or ketoconazole was assessed by the investigator as probably related, possibly related, or unrelated.
Statistical Analysis
Statistical analyses for safety were performed using SAS version 9.2 or higher (Cary, North Carolina). Statistical analyses for pharmacokinetic comparisons were performed using WinNonlin version 6.4 (Certara Inc. St. Louis, Missouri). The safety population was defined as all subjects who received at least one dose of study drug. The pharmacokinetic population was defined as all subjects who received study drug and for whom the pharmacokinetic profile could be adequately characterized. Pharmacokinetic steady-state parameters of mifepristone when administered alone (period 1, day 12) were compared to the steady-state parameters when administered with concomitant ketoconazole at steady state (period 2, day 17) to estimate the magnitude of the effect of ketoconazole on mifepristone. A linear mixed-effects model was used to estimate the geometric least squares mean ratios (GMRs) and associated 90% confidence intervals (CIs) from the pharmacokinetic parameters (log-transformed values of C max and AUC). Comparisons of the steady-state pharmacokinetic parameters were considered not clinically different if the 90% CIs around the GMR were within the standard bioequivalence interval of 80:125 [16]. Median differences in t max were computed using the Wilcoxon matched-pairs signed rank test for paired comparisons and the Mann–Whitney test for any unpaired comparisons; 90% CIs were computed using the Hodges–Lehmann method. Similar methods were used to compare GMRs and associated 90% CIs for the pharmacokinetic parameters of ketoconazole following multiple doses of ketoconazole (200 mg twice daily) plus mifepristone on day 17 (log-transformed C max and AUC0–12) to a single dose of ketoconazole (200 mg) on day − 1 (C max and AUCinf).
Sample size calculations determined that a sample size of 8 would achieve an 82% power to detect a mean of paired differences in AUC0–24 of 18,857 ng h/mL with an estimated standard deviation (SD) of 15,963 ng h/mL and a significance level of 0.05 using a two-sided paired t test. A prespecified sample size of 16 subjects was selected for this study in order to account for an approximate discontinuation rate of up to 50%.
Serum cortisol and plasma ACTH values were summarized with descriptive statistics (mean [SD] or median [range]). A Wilcoxon signed rank test was performed to examine the within-subject changes in cortisol and ACTH on day 17 of mifepristone plus ketoconazole compared with day 13 of mifepristone alone.
Results
Study Population
Overall, 16 healthy men (mean age 31.9 years [range 23–45 years]) were admitted to the study (Table 1); all received at least one dose of study drug and were included in the safety population. Two subjects discontinued mifepristone during period 1 because of AEs. Of the 14 remaining subjects who completed the study, one subject had notably high mifepristone concentrations at all time points (3.01 times the mean) and was excluded prior to any mifepristone analysis as an outlier. The reason for this apparent outlier was unknown. In the reference study (1200 mg mifepristone), 24 subjects (mean age 42.7 years [range 22–61] years, 75% men, mean body mass index 27.0 kg/m2) were enrolled, of which 22 completed the study.
Effects of Steady-State Ketoconazole on the Pharmacokinetics of Mifepristone and Metabolites
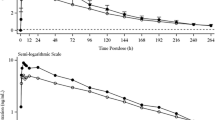
Mean trough concentrations of mifepristone and its metabolites were increasing to steady state on days 10–12 before ketoconazole was administered (Fig. 3). Plasma concentration–time curves for steady-state mifepristone and its metabolites before and after CYP3A4 inhibition with ketoconazole are presented in Fig. 3a–c; pharmacokinetic parameters are presented in Table 2. The pharmacokinetic profile following multiple doses of mifepristone alone showed that mifepristone was rapidly absorbed, with a median t max of 4.25 h (Fig. 3a). Following concomitant ketoconazole administration, concentrations of mifepristone were higher (day 17) than mifepristone administered alone (day 12). For mifepristone, the upper bound of the 90% CI for the calculated GMR of exposure exceeded 125% for C max and AUC0–24. The increase in mifepristone systemic exposure based on the GMRs was 28% for C max and 38% for AUC0–24 (Table 2), which was less than the 5-fold or higher increase recommended for consideration as a sensitive CYP3A4 substrate. For metabolites RU 42848 and RU 42633, the calculated GMRs and 90% CIs of exposure ratios were within the FDA’s standard 80:125 comparison interval, indicating that exposure to these metabolites was unaffected by ketoconazole. The upper bound of the 90% CI of the calculated GMR exceeded 125% for C max and AUC0–24 for the RU 42698 metabolite, indicating that concomitant administration with ketoconazole increased exposure to this metabolite (Table 2). Median t max of mifepristone was unaltered (4.25 h on day 17 and day 12). There were no significant differences in the medians of paired differences for t max between day 12 and 17 for mifepristone or any of the three metabolites. Half-life was not reported because the mean terminal elimination half-life for mifepristone and its metabolites exceeded 64 h. As such, the measurement of half-life over the 24-h dose interval would be unreliable.

Mean (SD) plasma trough concentrations of mifepristone (a) and its metabolites: RU 42633 (b), RU 42698 (c), and RU 42848 (d) on days 10, 11, and 12 (left portion of graphs), followed by mean plasma concentration–time profiles in the presence (day 17) and absence (day 12) of ketoconazole (right portion of graphs)
Pharmacokinetics of Mifepristone with Concomitant Ketoconazole Compared to Mifepristone 1200 mg Alone (Cross-Study Comparison)
Systemic exposure (C max and AUC) to mifepristone following multiple doses of mifepristone 600 mg plus ketoconazole 200 mg twice daily was less than the exposure to mifepristone following multiple doses of mifepristone 1200 mg alone under similar fed conditions reported in the reference study data (Table 3). The GMR of exposure to mifepristone was 85% and 87% of the exposure observed following the highest label dose of 1200 mg/day for C max and AUC0–24, respectively. The upper bounds of the 90% CIs for the calculated GMR of C max and AUC0–24 did not exceed the standard 125% interval limit (Table 3).
For the metabolites RU 42633 and RU 42848, the upper bounds of the 90% CIs were less than 125% for C max and AUC0–24, whereas for metabolite RU 42698, the upper bounds of the 90% CIs exceeded the standard interval for C max and AUC0–24.
Effects of Steady-State Mifepristone on the Pharmacokinetics of Ketoconazole
Systemic exposure to ketoconazole following multiple doses of ketoconazole 200 mg twice daily administered with concomitant mifepristone 600 mg once daily over 5 days (day 17) were compared with the exposure following a single 200-mg dose of ketoconazole given alone (day − 1). Mean predose trough concentrations of ketoconazole during repeated dosing days 14–17 (1353.07–1615.64 ng/mL) suggested that steady state for ketoconazole in the presence of mifepristone was achieved by approximately day 16.
The GMR of exposure for C max and AUC and their 90% CIs were all above 125%, indicating that ketoconazole exposure following steady-state twice-daily 200-mg ketoconazole plus concomitant steady-state mifepristone (day 17) was higher than ketoconazole exposure following single-dose 200-mg ketoconazole administered alone (day − 1) (Table 4). The increased exposure to ketoconazole with concomitant multiple doses of mifepristone vs single-dose ketoconazole alone was 2.53-fold higher for C max (GLSM 5224 vs 2067 ng/mL) and 3.65-fold higher for AUC (GLSM 37,653 vs 10,306 h ng/mL). Median t max was 3.0 h (range 1.0–8.0 h) following single-dose ketoconazole vs 4.33 h (range 3.0–4.4 h) following steady-state ketoconazole plus steady-state mifepristone.
Effects of Concomitant Ketoconazole and Mifepristone on Cortisol and ACTH
Mean serum cortisol and plasma ACTH levels increased relative to baseline (screening period) and were elevated above the upper limit of normal (ULN) starting on day 6 of mifepristone alone (period 1) and remained elevated during period 2 with the addition of ketoconazole (Fig. 4a, b). Serum cortisol and plasma ACTH levels decreased to within normal limits by study end in all but one and four subjects, respectively. Mean serum cortisol levels were significantly lower on day 17 compared to day 13 (35.1 vs 41.9 µg/dL; p = 0.0107) (Fig. 4a). Mean ACTH levels were higher on day 17 compared to day 13, but the differences were not statistically significant (178 vs 148 pg/mL; p > 0.05) (Fig. 4b).

Mean (SD) serum cortisol (a) and plasma ACTH (b). ULN upper limit of normal
Safety
At least one treatment-emergent AE was reported in nine subjects (56.3%) during administration of mifepristone alone (period 1) and in eight subjects (57.1%) during coadministration of mifepristone and ketoconazole (period 2). The AEs were categorized as mostly mild in severity; four moderate AEs occurred, including three during period 1 (hypertension, rash, and vomiting) and one during period 2 (headache). There were no serious AEs or events of adrenal insufficiency reported during the study.
Two participants were discontinued from the study during period 1. One subject with elevated blood pressure upon study entry was discontinued on day 6 after the 5th dose of mifepristone because of moderate hypertension. The AE was classified by the investigator to be possibly related to mifepristone; however, an alternate etiology of anxiety was also noted. One other subject was discontinued on day 11 after the 10th dose of mifepristone because of a moderate bilateral rash, which was classified by the investigator as related to mifepristone and resolved within 4 days without sequelae. Mildly elevated liver enzymes were reported for one subject during the mifepristone plus ketoconazole dosing period, starting approximately 1 day after the first dose of ketoconazole. During the concomitant dosing period, aspartate transaminase and alanine transaminase ranged from 45 to 76 U/L (normal range 0–37 U/L) and 85 to 126 U/L (normal range 6–45 U/L), respectively. Both returned to normal ranges during the washout period. There were no clinically meaningful changes in hematology, biochemistry, ECG, urinalysis findings, or orthostatic vital signs.
Discussion
This study was conducted to examine the pharmacokinetic effects following concomitant mifepristone and ketoconazole administration. Coadministration of mifepristone 600 mg daily plus ketoconazole 200 mg twice daily resulted in a mean increase in exposure to mifepristone of approximately 28% for C max and 38% for AUC0–24. The increased exposure was approximately 85% of the maximum exposure following the highest labeled dose of mifepristone (1200 mg) and less than the at least 5-fold increase needed to be considered a sensitive substrate [16]. Coadministration with ketoconazole had little effect on the exposure to mifepristone’s mono-demethylated (RU 42633) or di-demethylated (RU 42848) metabolites; exposure to the hydroxylated metabolite (RU 42698) increased by approximately 70%. The mechanisms for the increased exposure to this metabolite could not be determined from this study. While it is unknown what the clinical effect of the increased exposure to the RU 42698 metabolite would be, its lower affinity for the glucocorticoid receptor than mifepristone likely minimizes its clinical importance relative to the exposure to mifepristone and the other active metabolites.
As expected, the GLSM C max and interval AUC0–12 of ketoconazole following multiple doses of ketoconazole 200 mg twice daily plus mifepristone was substantially higher (2.53- and 3.65-fold) than the C max and AUCinf following a single dose of ketoconazole 200 mg. The increased interval AUC0–12 (day 17) vs AUCinf after a single dose (day − 1) is consistent with the nonlinear pharmacokinetics of ketoconazole, which has shown greater than expected increases in AUC following multiple dosing [23]. Factors contributing to the increased exposure to ketoconazole in this analysis could include inhibition of CYP3A4 by mifepristone and/or autoinhibition of CYP3A4 by ketoconazole.
Serum cortisol and plasma ACTH significantly increased over baseline with mifepristone alone and when combined with ketoconazole. These increases are in line with the competitive glucocorticoid receptor antagonistic effects of mifepristone. Similar increases in ACTH and cortisol have been observed with mifepristone administration in other healthy (non-CS) populations [12]. While ACTH remained elevated with the addition of ketoconazole, serum cortisol significantly decreased with concomitant ketoconazole compared with mifepristone alone, likely due to the inhibition of steroidogenesis by ketoconazole. However, it remains to be determined what the pharmacodynamic effects of concomitant ketoconazole and mifepristone would be in patients with baseline hypercortisolism and hypothalamic-pituitary-adrenal axis dysfunction.
AEs observed during the study were mostly mild or moderate in severity and were generally consistent with the known safety profiles of mifepristone and ketoconazole. Overall, similar rates of drug-related AEs were reported during period 1 and period 2, and there was no evidence that would suggest an increase in AEs under the conditions studied.
In order to optimize the possibility of detecting a potential drug–drug interaction between mifepristone and ketoconazole both compounds were administered with food to increase their absorption. The doses used for each compound in this study were within the recommended clinical doses for each agent [4, 20]; however, the median dose used for ketoconazole in the long-term off-label treatment of CS is higher (600 mg/day) [19]. The pharmacokinetic profile of mifepristone assessed at steady state before concomitant ketoconazole was generally consistent with the known pharmacokinetic profile [4]. Also, increases in cortisol and ACTH levels were seen, as expected, following repeated mifepristone administration during period 1. These findings suggest that the pharmacokinetic analysis provided an accurate assessment of the effects of ketoconazole on mifepristone.
There are several limitations of this study that must also be considered. Although multiple doses of ketoconazole were administered using the maximum recommended dose, the duration of concomitant treatment with ketoconazole (5 days) may not have been long enough to observe the full magnitude of inhibition by ketoconazole on mifepristone because of the long half-life of mifepristone. The rationale for shortening the duration of treatment with ketoconazole was based on its known hepatotoxicity potential. Likewise, the analysis of the effects of mifepristone on the pharmacokinetics of ketoconazole was also designed to limit exposure to ketoconazole, but as a result, the comparison of ketoconazole pharmacokinetics before and during concomitant mifepristone may have overestimated the contribution of mifepristone, by comparing a single dose vs multiple dosing. Of note, since the completion of this study, the FDA has advised using alternate strong CYP3A4 inhibitors, such as itraconazole or clarithromycin, over ketoconazole in clinical pharmacokinetic studies because of the risk of liver injury [24]. In the cross-study comparison, both studies included healthy subjects and used similar dosing conditions (fed state) and pharmacokinetic blood sampling schedules; however, the subject variability may have impacted the results. Furthermore, because the analysis was conducted in healthy subjects, the effects of concomitant dosing on the various aspects of clinical disease status are unknown, which limits the conclusions that can be made regarding definitive dosing recommendations.
On the basis of the results of this study, it is recommended that the dose of mifepristone be limited to 600 mg daily when coadministered with a strong CYP3A4 inhibitor like ketoconazole. However, because of mifepristone’s long half-life and nonlinear pharmacokinetic profile, with less than proportional increases in exposure with increasing doses, additional studies may be needed to determine if longer durations of concomitant administration and/or using mifepristone doses beyond 600 mg will produce exposure within the acceptable ranges. This is of clinical importance because of the dose-dependent effects at the glucocorticoid receptor, and an additional assessment of mifepristone efficacy and safety in patients with CS has noted trends toward improved efficacy at the higher dose ranges of mifepristone and with no discernable increase in AEs with increasing dose [25].
Conclusion
Together these data indicate increased pharmacokinetic exposure to mifepristone following mifepristone 600 mg combined with a strong CYP3A4 inhibitor, ketoconazole. The increase in systemic exposure was within the exposure bounds of the maximum 1200 mg dose of mifepristone. Dose adjustment of mifepristone may be required when coadministering mifepristone and ketoconazole.
References
Fleseriu M, Biller BM, Findling JW, Molitch ME, Schteingart DE, Gross C. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J Clin Endocrinol Metab. 2012;97:2039–49.
Heikinheimo O, Lahteenmaki PL, Koivunen E, et al. Metabolism and serum binding of RU 486 in women after various single doses. Hum Reprod. 1987;2:379–85.
Swahn ML, Wang G, Aedo AR, Cekan SZ, Bygdeman M. Plasma levels of antiprogestin RU 486 following oral administration to non-pregnant and early pregnant women. Contraception. 1986;34:469–81.
Corcept Therapeutics. Korlym® (mifepristone) 300 mg tablets [prescribing information]. http://www.korlym.com/docs/KorlymPrescribingInformation.pdf. Accessed 15 June 2017.
Jang GR, Wrighton SA, Benet LZ. Identification of CYP3A4 as the principal enzyme catalyzing mifepristone (RU 486) oxidation in human liver microsomes. Biochem Pharmacol. 1996;52:753–61.
Wu WN, McKown LA, Moyer MD, Johannsen TB, Takacs AR. In vitro metabolism of mifepristone (RU-486) in rat, monkey and human hepatic S9 fractions: identification of three new mifepristone metabolites. Xenobiotica. 1999;29:1089–100.
Heikinheimo O, Kontula K, Croxatto H, Spitz I, Luukkainen T, Lahteenmaki P. Plasma concentrations and receptor binding of RU 486 and its metabolites in humans. J Steroid Biochem. 1987;26:279–84.
He K, Woolf TF, Hollenberg PF. Mechanism-based inactivation of cytochrome P-450-3A4 by mifepristone (RU486). J Pharmacol Exp Ther. 1999;288:791–7.
Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–23.
Blasey CM, Block TS, Belanoff JK, Roe RL. Efficacy and safety of mifepristone for the treatment of psychotic depression. J Clin Psychopharmacol. 2011;31:436–40.
DeBattista C, Belanoff J, Glass S, et al. Mifepristone versus placebo in the treatment of psychosis in patients with psychotic major depression. Biol Psychiatry. 2006;60:1343–9.
Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology. 2006;31:628–36.
ClinicalTrials.gov. Mifepristone and eribulin in patients with metastatic triple negative breast cancer or other specified solid tumors. https://clinicaltrials.gov/ct2/show/NCT02014337?term=mifepristone+and+breast+cancer&rank=4. Accessed June 14, 2017.
ClinicalTrials.gov. A trial of nanoparticle albumin-bound paclitaxel (nab-paclitaxel, Abraxane®) with or without mifepristone for advanced, glucocorticoid receptor-positive, triple-negative breast cancer. https://clinicaltrials.gov/ct2/show/NCT02788981?term=mifepristone+and+breast+cancer&rank=3. Accessed June 14, 2017.
ClinicalTrials.gov. Enzalutamide and mifepristone in treating patients with metastatic hormone resistant prostate cancer. https://clinicaltrials.gov/ct2/show/NCT02012296. Accessed June 14, 2017.
US Food and Drug Administration. Guidance for industry. Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. http://www.fda.gov/downloads/Drugs/ucm292362.pdf. Accessed July 15, 2013.
Fitch WL, Tran T, Young M, Liu L, Chen Y. Revisiting the metabolism of ketoconazole using accurate mass. Drug Metab Lett. 2009;3:191–8.
Sonino N, Boscaro M, Paoletta A, Mantero F, Ziliotto D. Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients. Clin Endocrinol. 1991;35:347–52.
Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab. 2014;99:1623–30.
Janssen Pharmaceuticals Inc. Nizoral® tablets [prescribing information]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/018533s040lbl.pdf. Accessed 13 June 2017.
Zhao P, Ragueneau-Majlessi I, Zhang L, et al. Quantitative evaluation of pharmacokinetic inhibition of CYP3A substrates by ketoconazole: a simulation study. J Clin Pharmacol. 2009;49:351–9.
Daneshmend TK, Warnock DW, Ene MD, et al. Influence of food on the pharmacokinetics of ketoconazole. Antimicrob Agents Chemother. 1984;25:1–3.
Brass C, Galgiani JN, Blaschke TF, Defelice R, O’Reilly RA, Stevens DA. Disposition of ketoconazole, an oral antifungal, in humans. Antimicrob Agents Chemother. 1982;21:151–8.
US Food and Drug Administration. FDA advises against using oral ketoconazole in drug interaction studies due to serious potential side effects. https://www.fda.gov/Drugs/DrugSafety/ucm371017.htm. Accessed July 15, 2013.
Yuen KC, Williams G, Kushner H, Nguyen D. Association between mifepristone dose, efficacy, and tolerability in patients with Cushing’s syndrome. Endocr Pract. 2015;21:1087–92.
Acknowledgements
This study, the article processing charges, and the open access fee were funded by Corcept Therapeutics. The authors wish to thank Thaddeus Block, MD, of Corcept Therapeutics, for his contributions to study concept and data analysis and Dan Combs, BS, of Combs Consulting Service (Mountain View, California) for his pharmacokinetic data analysis, which was funded by Corcept Therapeutics. The authors also thank Diana Talag, ELS, of MedVal Scientific Information Services, LLC (Princeton, New Jersey), for editorial assistance. Support for this assistance was funded by Corcept Therapeutics. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Dat Nguyen is an employee of Corcept Therapeutics. Sarah Mizne is an employee of MedVal Scientific Information Services, LLC.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients for being included in the study. The protocol was approved by an independent investigational review board (IntegReview, Austin, Texas). This manuscript was prepared according to the International Society for Medical Publication Professionals’ “Good Publication Practice for Communicating Company-Sponsored Medical Research: the GPP3 Guidelines” and the International Committee of Medical Journal Editors’ “Uniform Requirements for Manuscripts Submitted to Biomedical Journals.”
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/D5BCF0602C731D7F.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nguyen, D., Mizne, S. Effects of Ketoconazole on the Pharmacokinetics of Mifepristone, a Competitive Glucocorticoid Receptor Antagonist, in Healthy Men. Adv Ther 34, 2371–2385 (2017). https://doi.org/10.1007/s12325-017-0621-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-017-0621-9