
Abstract
Introduction
To clarify the additional efficacy and safety benefits of 24 months’ treatment with the once-weekly formulation of teriparatide, which is currently used for 72 weeks.
Methods
This was a multicenter, open-label, single-arm study conducted in Japan. Subjects who were 65 years or older with prevalent vertebral fractures received once-weekly subcutaneous injection of 56.5 μg teriparatide for 24 months. The main outcome measure was percentage change from baseline in lumbar (L2–L4) BMD measured by dual-energy X-ray absorptiometry.
Results
A total of 189 subjects received at least one dose of the once-weekly formulation of teriparatide. Lumbar, femoral neck, and total hip BMD increased significantly compared with baseline at Weeks 24, 48, 72, and 104. In addition, significant increases in lumbar (+1.5%) and femoral neck (+0.8%) BMD were noted at Week 104 compared with Week 72. Significant increases from baseline in BMD for radius 1/10 were noted at Weeks 24 and 104. No substantial increases were noted in the cumulative incidences of new vertebral fracture and other types of fracture after Week 72. The safety profile seen in the first 72 weeks remained unchanged until 104 weeks.
Conclusion
The once-weekly formulation of teriparatide is effective and safe for the treatment of osteoporosis over 24 months. The limitation of this study is that this was an open-label, single-arm study.
Funding: Asahi Kasei Pharma Corporation.
Clinical Trial Registration: JapicCTI-132276.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Teriparatide is a peptide comprising the 34 amino acids of the biologically active N-terminal portion of human parathyroid hormone. It is the only osteoporosis drug currently available with an osteogenesis-promoting effect. Teriparatide increases bone mineral density (BMD) and improves bone structure, thereby exhibiting a powerful effect to inhibit fractures [1, 2]. A daily self-administered 20-μg formulation is available worldwide. In addition to the daily formulation, a once-weekly 56.5-μg formulation of teriparatide is also available in Japan and South Korea.
A placebo-controlled, 72-week, Teriparatide Once-Weekly Efficacy Research (TOWER) trial of the once-weekly formulation conducted in Japanese osteoporosis patients with a high risk of fractures showed that, compared with placebo, the formulation had a powerful fracture-inhibition effect, providing an 80% reduction in the relative risk of new vertebral fractures, and it increased lumbar BMD, total hip BMD, and femoral neck BMD by 6.7%, 3.1%, and 1.8%, respectively, from the baseline level [2].
Meanwhile, in response to confirmation of osteosarcoma in teriparatide-treated rats [3, 4], the periods for clinical trials were determined using the results of the study in animals as a reference. A limit of 24 months and 72 weeks, respectively, was placed on the duration of treatment with the daily formulation and the once-weekly formulation on the basis of the track records of treatment in clinical trials, giving rise to a difference in the duration of treatment. While it is desirable to make the once-weekly formulation of teriparatide available to patients for the same duration of treatment as that for the daily formulation, the efficacy and safety of the once-weekly formulation beyond 72 weeks have not yet been verified.
To clarify the additional efficacy and safety benefits of 24 months’ treatment with the once-weekly formulation of teriparatide, which is currently used for 72 weeks, a decision was made to conduct a 24-month open-label study in Japanese patients with primary osteoporosis with a high risk of fractures. The possibility of an increased risk of developing osteosarcoma associated with the extended duration of treatment was taken into consideration before conducting this study. The onset of osteosarcoma is believed to be dependent on the total amount of teriparatide administered [5]. With the once-weekly formulation, the weekly dose of teriparatide is 56.5 μg, which is less than half of that of 140 μg with the daily formulation. Moreover, the incidence of osteosarcoma among patients who received treatment with the daily formulation was not more than the spontaneous incidence among the general population [6]. Thus, it was concluded that there would be no increase in the risk of developing osteosarcoma even if the duration of treatment with the once-weekly formulation were extended to 24 months, leading to a decision to conduct the study.
Methods
Study Design
This study was a multicenter, open-label, single-arm study conducted in Japan (JapicCTI-132276). The subjects received once-weekly subcutaneous injection of 56.5 μg teriparatide for 24 months (104 weeks) and standard therapy that included daily oral doses of calcium (610 mg), vitamin D (400 IU), and magnesium (30 mg) (New Calcichew D3; Daiichi Sankyo Healthcare Co., Ltd., Tokyo, Japan).
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice (GCP). Moreover, institutional review board (IRB) approval of the protocol was in place before the study at each of the 25 study sites.
Subjects
The eligibility criteria established for study subjects were as follows: man or woman aged 65 years or older; able to walk independently; diagnosis of primary osteoporosis based on the diagnostic criteria for primary osteoporosis (FY2012 revised version) by the Japanese Society for Bone and Mineral Research [7]; at least one but no more than five prevalent fractures between the fourth thoracic vertebra (Th4) and the fourth lumbar vertebra (L4); and a mean BMD of the second through fourth lumbar vertebrae (L2–L4) of less than 80% of the young adult mean (YAM) at the time of enrollment. Patients diagnosed with secondary osteoporosis, patients with a non-osteoporotic disease that causes decreased bone mass, and patients with any X-ray findings that affect the assessment of lumbar BMD by dual energy X-ray absorptiometry (DXA) were excluded. Also excluded were patients with any of the following conditions and those judged by the investigator as unsuitable for participation in the clinical study: a serum calcium level of 11.0 mg/dl or above; a malignant bone tumor or a metastatic bone tumor; previous radiation therapy affecting the bone or otherwise at high risk of developing osteosarcoma; or a serum alkaline phosphatase (ALP) level more than double the standard level. Patients who had received treatment with teriparatide or an anti-receptor activator of nuclear factor kappa B ligand (RANKL) antibody in the past, bisphosphonate within 52 weeks before treatment commencement, or any other osteoporosis drug within 8 weeks before treatment commencement were also excluded.
Efficacy Endpoints
The primary endpoint selected was the percentage change from baseline in L2–L4 BMD at week 104. The secondary endpoints were the percentage changes in total hip BMD and femoral neck BMD and X-ray-confirmed new vertebral fractures and clinical fractures. Other variables assessed included the time profiles of bone turnover markers (bone formation and bone resorption markers). In addition, radial BMDs (distal 1/3, 1/6, and 1/10 sites of the radius) were measured with peripheral dual-energy X-ray absorptiometry (DXA) at two study sites. The differences in BMD values between Week 72 and Week 104 were also examined.
Efficacy Measures
BMDs of the lumbar spine, femur, and radius were measured by DXA at screening, baseline, and Weeks 24, 48, 72, and 104. The lumbar and femoral BMDs were measured with a Discovery, Explorer, Delphi, or QDR4500 (Hologic, Bedford, MA) device, which was calibrated before each test for precision control with a lumbar spine phantom attached to the device. For external quality control, specialists checked QC sheets from all study sites every month and then maintenance was performed if needed. The radial BMDs were measured at two study sites equipped with a peripheral DXA. Both study sites used a DCS-600EXV (Hitachi Ltd., Tokyo, Japan) to perform the measurements. The lumbar and femoral BMD measurements were centrally analyzed by a BMD analysis institution and then assessed independently by two members of a Bone Mass-assessment Committee (HH, TeS). When the two members differed in their assessments, the Bone Mass-assessment Committee considered the case to render an assessment that was considered final. The radial BMD measurements were centrally analyzed by the BMD analysis institution and then assessed through a consultation in the Bone Mass-assessment Committee by the two members of the committee to make an assessment that was considered final.
To assess morphological vertebral fractures, X-ray images of the lumbar/thoracic vertebrae were taken at screening and baseline and at 24, 48, 72, and 104 weeks after. A Fracture-assessment Committee (TeN, HK, MI, TeS) assessed the vertebrae from Th4 through L4 by a semi-quantitative method (SQ) [8] and a quantitative method (QM) [9, 10]. A new vertebral fracture was defined as an increase of one grade or more of a vertebral body that was normal (SQ grade 0) at baseline combined with a 20% or greater reduction in height at either the anterior edge, center, or posterior edge of the vertebral body. A clinical fracture was defined as a fracture associated with clinically apparent symptoms such as pain that could be confirmed by X-ray imaging or magnetic resonance imaging (MRI). Clinical fractures were confirmed by the investigator at each of the study sites. A fragility fracture was defined as a clinical fracture that the investigator judged was not induced by a large external force.
To measure bone turnover markers, blood and urine samples were collected at baseline and before the study drug was administered at Weeks 4, 12, 24, 48, 72, and 104. Samples were stored either in a refrigerator or a freezer, depending on the type of marker to be measured, before measurement by a validated central laboratory (LSI Medience Corporation, Tokyo, Japan). Serum osteocalcin (OC) was measured by immunoradiometric assay (BGP-IRMA; LSI Medience Corporation, Tokyo, Japan); serum procollagen type I amino-terminal propeptide (P1NP) was measured by radioimmunoassay (Fujirebio Inc., Tokyo, Japan); and urinary cross-linked N-telopeptide of type I collagen (NTX) was measured by enzyme-linked immunoassay (Alere Medical Co., Ltd., Tokyo, Japan).
Adverse Events
Safety was assessed collectively on the basis of adverse events, including serious adverse events, and adverse events leading to study discontinuation. The subjects underwent medical examinations and regular blood tests, blood chemistry tests, urinalyses, and vital sign measurements. The investigators reported adverse events, which were coded to the preferred terms in the Medical Dictionary for Regulatory Activities (MedDRA, version 16.0).
Sample Size
To summarize the percentage change from baseline in lumbar (L2–L4) BMD data using descriptive statistics, it was adequate that the 95% confidence interval in this study was of a similar degree to that in the TOWER trial. On the basis of the sample size and the rate of discontinuations in the population used for lumbar BMD analysis in the TOWER trial, a target sample size of 175 subjects was selected to commence treatment in this study. In the TOWER trial [2], the population in the active drug group analyzed for lumbar BMD included 107 subjects at Week 72. Thus, to ensure a similar degree of 95% confidence interval (12.6–13.4) in this study, approximately 110 subjects were needed at Weeks 72 and 104. Given that, in the TOWER trial, the rate of discontinuations was 20% through Week 24, and 5.5% of the subjects discontinued the study over each 24-week period thereafter, the overall rate of discontinuation in this study was expected to reach 37%. Therefore, it was determined that 175 subjects were needed at the start of treatment to ensure the availability of 110 subjects at week 104.
Statistical Analysis
Efficacy was analyzed using the full analysis set (FAS). The FAS included all subjects who received at least one dose, except those who had any GCP deviation, those confirmed to have no primary osteoporosis, and those for whom no post-treatment efficacy data were available.
Percentage changes from baseline in L2–L4 BMD, the primary endpoint, were summarized over time to determine the 95% confidence interval for the percentage change. The total hip, femoral neck, and radial (distal 1/3, 1/6, and 1/10 sites of the radius) BMDs were also summarized in a similar manner. Cumulative incidences of fractures at each time point were estimated by the Kaplan–Meier method. Percentage changes from baseline in bone turnover markers were summarized over time. A comparison between baseline and each time point or between Week 72 and Week 104 for BMD was performed by the paired t test. A comparison between baseline and each time point for bone turnover markers was performed by the Wilcoxon rank sum test. The relationships between the BMD absolute change category and the incident nonvertebral fracture number were assessed by the Chi square test.
Safety was analyzed using the population that included all subjects who received at least one dose. Incidences of each adverse event were summarized by onset time. Statistical analyses were performed using SAS version 9.1 or 9.4 (SAS Institute, Cary, NC, USA).
Results
Subjects
Of 317 subjects who gave informed consent at 25 study sites throughout Japan, 189 were enrolled in the study (Fig. 1). All enrolled subjects received at least one dose of teriparatide, but one of the subjects was excluded from the FAS because of a non-osteoporotic disease that causes decreased bone mass that became known after treatment commencement. The 188 subjects in the FAS had a mean age of 73.9 years and T scores of −3.1 for the lumbar spine, −2.5 for the total hip, and −3.1 for the femoral neck. Subjects in the FAS who had one prevalent vertebral fracture accounted for 46.8%, representing the largest subgroup (Table 1). Meanwhile, as a result of differences in the assessment of prevalent vertebral fractures between the investigator and the special committee, 23.4% of subjects were considered to have no prevalent vertebral fracture (the data which reflected the decision of the special committee was used in the analyses).

Disposition of patients
BMD
The percentage change from baseline in lumbar (L2–L4) BMD was 8.4% (95% confidence interval [CI], 7.4–9.4) at Week 72 and 9.9% (95% CI 8.7–11.1) at Week 104 (Fig. 2). The lumbar BMD increased significantly from baseline at every evaluation time point, with the increase persisting up to 104 weeks (24 months). After Week 72, the mean percentage change increased significantly by 1.5% from Weeks 72 to 104. The increase from baseline in total hip BMD was 2.7% (95% CI 2.1–3.3) at Week 72 and 2.8% (95% CI 2.1–3.4) at Week 104, and that in femoral neck BMD was 2.5% (95% CI 1.7–3.3) at Week 72 and 3.3% (95% CI 2.5–4.2) at Week 104, showing a significant increase from baseline at every body site and every evaluation time point. After Week 72, the mean percentage change of BMD for femoral neck increased significantly by 0.8% from Weeks 72 to 104. In addition, the percentage change from baseline in radial BMD was 1.4% (95% CI −2.4 to 5.3) at Week 72 and 2.3% (95% CI −1.0 to 5.6) at Week 104 at the distal 1/3 radius; 1.2% (95% CI −2.0 to 4.3) and 2.6% (95% CI −0.1 to 5.3), respectively, at the distal 1/6 radius; and 1.5% (95% CI −1.3 to 4.3) and 2.4% (95% CI 0.4–4.3), respectively, at the distal 1/10 radius, showing a significant increase from baseline at the distal 1/10 radius at Weeks 24 and 104.

Mean percentage change from baseline in BMD. a Lumbar spine (L2–L4), b total hip, c femoral neck, d distal radius. Bars indicate 95% confidence interval. Paired t test (compared with baseline) *p < 0.05; **p < 0.01
Fractures and Bone Turnover Markers
The cumulative incidence of new vertebral fractures estimated by the Kaplan–Meier method was 4.8% at Week 104 (Table 2). Moreover, the cumulative incidences of clinical fractures, of clinical vertebral fractures, and of clinical fragility fractures at Week 104 were 10.0%, 4.6%, and 6.9%, respectively. The cumulative incidences of nonvertebral fractures and of clinical nonvertebral fragility fractures at Week 104 were 6.1% and 3.6%, respectively, and they did not increase from Week 72 to Week 104.
The serum OC level increased significantly from baseline at Week 4 of treatment, with the increase sustained up to Week 12. It declined through to Week 48, but the increase from baseline remained significant until week 104, showing no great fluctuations (Fig. 3). The serum P1NP level increased significantly from baseline at Week 4 of treatment and then declined through to Week 48. Thereafter, the level remained below baseline up to Week 104. The urinary NTX level increased significantly from baseline at Week 12 of treatment, but it showed no substantial changes after treatment commencement.

Mean percentage change from baseline in bone turnover markers. a s-OC, b s-P1NP, and c u-NTX. Bars indicate 95% confidence interval. Wilcoxon rank sum test (compared with baseline) *p < 0.05; **p < 0.01
Treatment Response
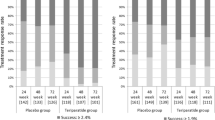
The proportion of subjects with a lumbar and femoral neck T score of −2.5 or greater increased over time from Weeks 0 to 104 (Fig. 4). Overall, 49.3% of the subjects were confirmed to have achieved a lumbar BMD T score of greater than −2.5 at Week 104. The incidence of nonvertebral fractures was 1.6% (1/64) in subjects with lumbar BMD T scores of −2.5 or greater; 6.1% (4/66) in subjects with T scores less than −2.5; 2.4% (1/41) in subjects with femoral neck BMD T scores of −2.5 or greater; and 6.5% (6/93) in those with T scores less than −2.5. Although the data showed no significant differences, the incidence of nonvertebral fractures tended to be lower in those with a T score of −2.5 or greater.

Proportion of subjects in the categories defined by the BMD of the young adult mean value in lumbar and femoral neck at the time of measurement. Numbers in parentheses show the number of subjects
Adverse Events
A total of 181 subjects (95.8%) experienced some kind of adverse events (Table 3). Serious adverse events occurred in 27 subjects (14.3%), but none was fatal. Particularly common adverse events were nasopharyngitis (76 subjects, 40.2%), nausea (69 subjects, 36.5%), vomiting (42 subjects, 22.2%), headache (37 subjects, 19.6%), malaise (33 subjects, 17.5%), contusion (30 subjects, 15.9%), abdominal discomfort (25 subjects, 13.2%), and dizziness (23 subjects, 12.2%), showing little difference from the incidence trend of adverse events in the TOWER trial [2].
The incidence of adverse events by time interval was similar across all intervals, and there were no adverse events that occurred at an increased rate after Week 72 (Table 3). No specific serious adverse events occurred at a high incidence. Adverse events leading to discontinuation occurred in 33 subjects (17.5%), with such events occurring in 18 subjects (9.5%) within the first 24 weeks, in 8 subjects (4.9%) after Week 24 through Week 48, in 6 subjects (4.0%) after Week 48 through Week 72, and in 2 subjects (1.4%) after Week 72, showing a higher trend in the initial phase of the study.
Discussion
The purpose of this study was to clarify the additional efficacy and safety benefits of 24 months’ treatment with the once-weekly formulation of teriparatide, which is currently used for 72 weeks. New results obtained from this study were an increasing trend in BMDs that still persisted after Week 72 and the absence of a trend for a marked increase in the incidence of fractures. With respect to safety, there were no new adverse events that had not been seen previously with the once-weekly formulation of teriparatide, showing that the safety profile seen in the first 72 weeks remained unchanged until Week 104.
Since treatments for osteoporosis are long term, it is important to establish a goal as a way of raising patients’ awareness about continuing treatment. Presently, there is a proposed treatment goal that includes BMD as one of its candidate indices [11]. BMD measurements, along with the confirmatory diagnosis of osteoporosis, are useful for monitoring the course of treatment. The risk of fracture is linked to the level of BMD decreases, with T score less than −2.5SD of the young adult mean (YAM) used globally as a threshold for osteoporosis [7, 12]. Reports to date have shown that once the T score for the lumbar spine [13] or femoral neck [14] reaches a level greater than −2.5, the incidence of fractures decreases significantly thereafter.
In the present study, the proportion of subjects with a lumbar spine (L2–L4) and femoral neck T score of −2.5 or greater increased over time from Weeks 0 to 104, with about half of the subjects confirmed to have achieved a lumbar BMD T score of greater than −2.5 at Week 104. Although the data showed no significant differences, the incidence of fractures tended to be lower in those with a T score greater than −2.5. These results indicate that extending the once-weekly treatment with teriparatide to 104 weeks can free a large number of patients from low BMD and, as a result, may also reduce the risk of fractures.
In addition, this study confirmed that the distal 1/10 radius BMD increased significantly at weeks 24 and 104, and the distal 1/3 and 1/6 radius BMDs showed an increasing trend relative to baseline at week 104. In a 24-month study of the daily formulation of teriparatide, the BMD of the radius shaft, which consists mainly of cortical bone, decreased significantly at the end of the study, and the distal radius BMDs also showed a decreasing trend [1], thus the effect on radial BMDs between the once-weekly and daily formulations may be different. This difference may be explained by the difference in mechanism of inducing bone formation between the once-weekly formulation and the daily formulation. As shown in the TOWER trial [2], the increase from baseline in the serum OC level remained over the study period, and the urinary NTX level showed no substantial changes after treatment commencement, suggesting that the once-weekly formulation of teriparatide induces bone formation without promoting bone resorption. A study in mice has shown that bone formation took place predominantly by remodeling with a high frequency of teriparatide administration and by both remodeling and mini-modeling with a low frequency of administration [15]. Bone formation by modeling involves little increase in bone resorption, thus resulting in no increase in cortical porosity, while daily treatment, which increases bone formation predominantly by remodeling, has been reported to increase the porosity of the radius in association with the activation of remodeling [16]. Increased cortical porosity is associated with daily, not weekly, administration of equivalent doses of teriparatide in rabbits [17]. The fact that the daily formulation markedly decreases the BMD of the radius shaft, which consists of more cortical bone than the distal end, suggests that the difference in effect on the porosity of cortical bone may be related to the difference in changes of radial BMD.
Furthermore, as shown in Fig. 2d, in this study, the timing of the start of the BMD increase at the distal end varied depending on the part of radius. While the distal 1/3 radius BMD showed no increasing trend at Week 24, the distal 1/6 and 1/10 radius BMDs showed an increasing trend relative to the baseline level after 24 weeks. Similar to the results from the present study, in the prior study that measured radial BMDs in subjects treated with the once-weekly formulation for 24 weeks, the distal 1/10 radius BMD increased significantly relative to the baseline level at 24 weeks after treatment commencement, but the distal 1/3 radius BMD showed no increase [18]. Differences in bone tissue among different parts of the radius may have an effect on the difference in the timing of this increase. Compared with the distal 1/3 radius, the distal 1/10 radius has more cancellous bone. Since bone turnover is higher in cancellous bone than in cortical bone [19], the effect of increasing BMD was seen earlier in the distal 1/10 radius.
Given that the once-weekly formulation tends to increase lumbar and femoral BMDs, as well as radial BMD, measurements of the early increases in the distal 1/10 radius BMDs are useful for raising awareness in patients in hospitals that are not equipped to measure lumbar spine and femoral BMDs. Moreover, the delayed increase in BMD in the distal 1/3 radius, which consists of more cortical bone than the distal 1/10 radius, may explain the absence of increases in the incidence of new fractures after Week 72 in nonvertebral bones, which consist mainly of cortical bone (Table 2). Long-term treatment with the once-weekly formulation of teriparatide may be effective in inhibiting fractures in the radius and other nonvertebral bones, which consist mainly of cortical bone.
The limitations of this study include the fact that it was a single-arm, open-label study and the fact that the primary endpoint was the percentage change of lumbar BMD from baseline. Nonetheless, the previous TOWER study showed that 83% (Freedman’s method) of vertebral fractures could be explained by changes in lumbar BMD [20], thus demonstrating that an increase of lumbar BMD is sufficient to indicate the treatment effect of the once-weekly formulation of teriparatide. Additionally, compared with the TOWER trial, which was conducted as a randomized controlled trial before this study, there were no substantial differences in subject characteristics and in efficacy and safety through Week 72 between this study and TOWER trial, and the study operations, including the assessments of bone mass and fractures, were properly carried out, thus allowing a comparison of results obtained through Week 72, as well as interpretations of effects after Week 72.
Conclusions
Up to now, there has been no experience in using the once-weekly formulation beyond 72 weeks. In this study, significant increases in lumbar and femoral neck BMDs were noted even after 72 weeks, and there was no trend of a marked increase in the incidence of fractures, indicating that extending the duration of teriparatide treatment from 72 weeks to 24 months can be expected to further maintain the inhibitory effect on fractures. With respect to safety, the safety profile seen in the first 72 weeks remained unchanged until Week 104. Therefore, the results indicate that the once-weekly formulation is useful for the treatment of osteoporosis over 24 months.
References
Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–41.
Nakamura T, Sugimoto T, Nakano T, et al. Randomized teriparatide [human parathyroid hormone (PTH) 1–34] once-weekly efficacy research (TOWER) trial for examining the reduction in new vertebral fractures in subjects with primary osteoporosis and high fracture risk. J Clin Endocrinol Metab. 2012;97:3097–106.
Vahle JL, Sato M, Long GG, et al. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1–34) for 2 years and relevance to human safety. Toxicol Pathol. 2002;30:312–21.
Watanabe A, Yoneyama S, Nakajima M, et al. Osteosarcoma in Sprague-Dawley rats after long-term treatment with teriparatide (human parathyroid hormone (1–34)). J Toxicol Sci. 2012;37:617–29.
Vahle JL, Long GG, Sandusky G, Westmore M, Ma YL, Sato M. Bone neoplasms in F344 rats given teriparatide [rhPTH(1–34)] are dependent on duration of treatment and dose. Toxicol Pathol. 2004;32:426–38.
Capriani C, Irani D, Bilezikian JP. Safety of osteoanabolic therapy: a decade of experience. J Bone Miner Res. 2012;27:2419–28.
Soen S, Fukunaga M, Sugimoto T, et al. Diagnostic criteria for primary osteoporosis: year 2012 revision. J Bone Miner Metab. 2013;31:247–57.
Genant HK, Wu CY, van Kuijk C, Nevitt MC. Vertebral fracture assessment using a semiquantitative technique. J Bone Miner Res. 1993;8:1137–48.
Tetsuro I, Kichizo Y, Hideaki T. Diagnostic criteria for osteoporosis: evaluation method of degree of bone mass reduction and vertebral body deformation by spinal plain radiograph. Ministry of Health and Welfare, Health Sciences Research Grant, Silver Science Research, FY1989 Report. 1990;118–9.
Hayashi Y, Kushida K, Kitazawa A, et al. Measurement of vertebral body dimensions of the thoracic and lumbar spines of 242 healthy women. J Bone Miner Metab. 1998;16:27–33.
Cummings SR, Cosman F, Lewiecki EM, et al. Goal-directed treatment for osteoporosis: a progress report from the ASBMR-NOF Working Group on goal-directed treatment for osteoporosis. J Bone Miner Res. 2017;32:3–10.
World Health Organization. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. WHO technical report series. Geneva: WHO; 1994.
Kuroda T, Shiraki M, Shiraki Y, Tanaka S. The importance of absolute bone mineral density in the assessment of antiresorptive agents used for the prevention of osteoporotic fractures. J Clin Densitom. 2012;15:392–8.
Shiraki M, Ueda S, Sugimoto T, Kuroda T, Nakamura T. Treatment responses with once-weekly teriparatide therapy for osteoporosis. Osteoporos Int. 2016;27:3057–62.
Yamamoto T, Hasegawa T, Sasaki M, et al. Frequency of teriparatide administration affects the histological pattern of bone formation in young adult male mice. Endocrinology. 2016;157(7):2604–20.
Macdonald HM, Nishiyama KK, Hanley DA, Boyd SK. Changes in trabecular and cortical bone microarchitecture at peripheral sites associated with 18 months of teriparatide therapy in postmenopausal women with osteoporosis. Osteoporos Int. 2011;22:357–62.
Zebaze R, Takao-Kawabata R, Peng Y, et al. Increased cortical porosity is associated with daily, not weekly, administration of equivalent doses of teriparatide. Bone. 2017;99:80–4.
Urushibara N, Kato N, Adachi R, et al. Once-weekly teriparatide increases bone mineral density in the distal 1/10 radius but not in the distal 1/3 radius. Springerplus. 2014;3:238–43.
Parfitt AM. Misconceptions (2): turnover is always higher in cancellous than in cortical bone. Bone. 2002;30:807–9.
Tanaka S, Kuroda T, Sugimoto T, Nakamura T, Shiraki S. Changes in bone mineral density, bone turnover markers, and vertebral fracture risk reduction with once weekly teriparatide. Curr Med Res Opin. 2014;30:931–6.
Acknowledgements
The authors would like to thank the investigators and clinical sites in Japan that participated in this study. The study, the article processing charges, and the Open Access fee were sponsored and funded by the Asahi Kasei Pharma Corporation, Tokyo, Japan. English editing was performed by William D. Stenson of FORTE Science Communications, and funded by Asahi Kasei Pharma Corporation. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published. All authors contributed equally to this work. Toshitsugu Sugimoto, Masataka Shiraki, Masao Fukunaga, Hiroshi Hagino, Teruki Sone, Tetsuo Nakano, Hideaki Kishimoto, Masako Ito, Hideki Yoshikawa, Mitsukazu Kishida, and Toshitaka Nakamura designed the study and contributed to data collection. Mitsukazu Kishida and Chika Irie contributed to data analysis and wrote the initial draft of the manuscript. Toshitsugu Sugimoto and Teruki Sone contributed to interpretation of data and assisted in the preparation of the manuscript. All other authors critically reviewed the manuscript. The final version of the manuscript was approved by all authors.
Disclosures
Toshitsugu Sugimoto has received research grants from Astellas Pharma, Eisai, Ono Pharmaceutical, Daiichi-Sankyo, Chugai Pharmaceutical, and Eli Lilly Japan and consulting and/or lecture fees from Asahi Kasei Pharma and Pfizer. Masataka Shiraki has received consulting fees from Asahi Kasei Pharma, MSD, and Teijin Pharma and received lecture fees from Chugai Pharmaceutical, Daiichi-Sankyo, Eisai, Eli Lilly Japan, Ono Pharmaceutical, and Pfizer. Masao Fukunaga has received consulting fees from Asahi Kasei Pharma and lecture fees from Daiichi-Sankyo and MSD. Hiroshi Hagino has received research grants, consulting fees, and/or lecture fees from Asahi Kasei Pharma, Astellas Pharma, Chugai Pharmaceutical, Daiichi-Sankyo, Eisai, Eli Lilly Japan, Mitsubishi Tanabe Pharma, Mochida Pharmaceutical, MSD, Ono Pharmaceutical, Pfizer, and Taisho Toyama Pharmaceutical. Teruki Sone has received research grants from Asahi Kasei Pharma, Astellas Pharma, Daiichi-Sankyo, Taisho Toyama Pharmaceutical, Takeda Pharmaceutical, Pfizer, and Teijin Pharma, and consulting fees from Takeda Pharmaceutical. Tetsuo Nakano has received consulting fees from Asahi Kasei Pharma, Chugai Pharmaceutical, Daiichi-Sankyo, and Teijin Pharma. Hideaki Kishimoto has received consulting fees from Asahi Kasei Pharma, Daiichi-Sankyo, EA Pharma, and Eisai. Masako Ito has received research grants from Asahi Kasei Pharma, Astellas Pharma, Chugai Pharmaceutical, Daiichi-Sankyo, MSD, and Ono Pharmaceutical, and consulting fees from Asahi Kasei Pharma and Ono Pharmaceutical. Hideki Yoshikawa has no competing interest. Mitsukazu Kishida is an employee of Asahi Kasei Pharma. Chika Irie is an employee of Asahi Kasei Pharma. Toshitaka Nakamura has received consulting fees from Asahi Kasei Pharma, Amgen, Chugai Pharmaceutical, Daiichi-Sankyo, MSD, Taisho Toyama Pharmaceutical, and Teijin Pharma.
Compliance with Ethics Guidelines
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice (GCP). Moreover, institutional review board (IRB) approval of the protocol was in place before the study at each of the study sites. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients for being included in the study.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available because of the sponsor’s policy as data sharing is not required in Japan.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/6598F0605ECE2FBC.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sugimoto, T., Shiraki, M., Fukunaga, M. et al. 24-Month Open-Label Teriparatide Once-Weekly Efficacy Research Trial Examining Bone Mineral Density in Subjects with Primary Osteoporosis and High Fracture Risk. Adv Ther 34, 1727–1740 (2017). https://doi.org/10.1007/s12325-017-0568-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-017-0568-x