
Abstract
Introduction
The superiority of tiotropium/olodaterol is demonstrated in improvement of lung function, dyspnea, lung hyperinflation, and quality of life compared with either monotherapy in patients with chronic obstructive pulmonary disease (COPD). Japanese Respiratory Society Guidelines for COPD management include improvement of exercise tolerance and daily physical activity as the treatment goals; however, there is limited evidence in Japanese patients with COPD.
Methods
A protocol is developed for the VESUTO® study that investigates the efficacy of tiotropium/olodaterol fixed-dose combination (FDC) compared with tiotropium alone on inspiratory capacity (IC, volume from functional residual capacity to total lung capacity), exercise capacity, and daily physical activity in Japanese patients with COPD.
Results
A total of 180 Japanese patients with COPD, aged ≥40 years will be enrolled into the double-blind, multicenter, active-controlled, crossover study (NCT02629965) and will be randomized to receive either tiotropium/olodaterol FDC or tiotropium for 6 weeks each [two puffs via RESPIMAT® (Boehringer Ingelheim, Ingelheim, Germany) inhaler in the morning]. The primary endpoint is IC at rest measured at 60 min post-dose after 6 weeks treatment. The secondary endpoints include the 6-min walk distance (6MWD) at 90 min post-dose and physical activity measured by the activity monitor in the last 2 weeks of the 6-week treatment periods. Lung function tests will also be assessed after 6 weeks treatment. A mixed-effects model repeated measures approach will be used for the primary and secondary endpoints.
Conclusion
The VESUTO® study is the first randomized interventional study to investigate exercise capacity (6MWD) and physical activity measured by a 3-axis accelerometer in Japanese patients with COPD. The study could provide additional evidence of long-acting muscarinic antagonist (LAMA) + long-acting β2-agonist (LABA) combination therapy on patients’ physical activities as well as lung function.
Trial registration
ClinicalTrials.gov: NCT02629965 (registered on December 1, 2015).
Funding
The VESUTO study was funded by Nippon Boehringer Ingelheim Co., Ltd., Tokyo, Japan.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic obstructive pulmonary disease (COPD), a leading cause of morbidity and mortality, is characterized by persistent airflow limitation that is usually progressive [1]. The prevalence of COPD in Japan is reported to be 8.6%, which is comparable to the global prevalence [2]. In Japan, approximately 5.3 million individuals ≥40 years old and 2.1 million individuals ≥70 years old are estimated to be affected by COPD [3]. Bronchodilators are recommended as first-line therapy in symptomatic patients with airflow limitation, and are fundamental to the management of stable COPD [4, 5]. Long-acting muscarinic antagonist (LAMA) and long-acting β2-agonist (LABA) combination therapy is recommended if symptoms persist despite treatment with bronchodilator monotherapy [5]. Fixed-dose combinations (FDCs) of LAMA + LABA can maximize the bronchodilator response without increasing the dose of individual components by LABA-mediated stimulation of β2-adrenergic receptors, LAMA-mediated inhibition of acetylcholine action at muscarinic receptors, and signaling cross-talk-induced synergistic bronchodilation effects [6, 7]. Further, the safety of LAMA + LABA combination therapies and monotherapies is comparable [8, 9].
Tiotropium/olodaterol FDC is a LAMA + LABA FDC approved for use in many countries, including Japan, the European Union, and the United States. Numerous randomized, double-blind, placebo-controlled, phase III studies (with durations of 6, 12, and 52 weeks), including parallel-group and crossover studies, have shown that treatment with tiotropium/olodaterol FDC (5/5 and/or 2.5/5 µg) significantly improved lung function [10,11,12], quality of life [10, 13], and transition dyspnea index [13] compared with either monotherapy in patients with moderate-to-very severe COPD [Global Initiative for Chronic Obstructive Lung Disease (GOLD), grades 2–4]. In particular, in the Japanese subpopulation of replicated phase III studies (TONADO® 1 and 2), tiotropium/olodaterol FDC (5/5 µg) was superior to either monotherapy for lung function and St George’s Respiratory Questionnaire (SGRQ) total score at 24 weeks, being consistent with results of the overall population [14].
Also, in several phase III studies, tiotropium/olodaterol FDC showed a significant improvement/reduction in lung hyperinflation [an increase in inspiratory capacity (IC)] after 6 or 12 weeks treatment compared with either monotherapy or placebo [15, 16]. However, no Japanese patients with COPD were included in these studies. Importantly, Japanese patient characteristics such as age, body mass index, smoking habits, maintenance treatment usage, and environmental exposures differ from those of Western patients [17, 18]. Therefore, the VESUTO® study has been planned to investigate the effect of tiotropium/olodaterol FDC and tiotropium monotherapy on IC as an index of lung hyperinflation in Japanese patients with COPD, because lung hyperinflation is one of the main factors for dyspnea, fatigue, and low exercise capacity [19, 20]. The 6-min walk distance (6MWD) is a validated measure of exercise capacity and correlates well with IC [21], responds to interventions with bronchodilators [22], and predicts morbidity and mortality [23]. Reduction in hyperinflation (increase in IC) with tiotropium/olodaterol FDC is considered to be associated with improvement in exercise capacity [15, 16] and possibly in physical activity [24]. Therefore, it is expected that improvement of symptoms resulting from reduction of hyperinflation will contribute to increasing patients’ exercise capacity and their daily activity [25]. Physical activity levels could be a new therapeutic target for COPD management [26], because low physical activity is associated with poor prognosis and is the most influential factor affecting all-cause mortality in COPD [27, 28], ranking as the third most important factor associated with death caused by lifestyle-related diseases in Japan [29]. Assessment of physical activity in the earliest stages of COPD is associated with better prognosis, as it helps maintain best achievable physical activity [27, 28]. Nonetheless, Japan is known for taking active measures to improve the health of its population [29]. Guidelines for the diagnosis and treatment of COPD issued by the Japanese Respiratory Society specifically include improvement in exercise capacity and daily physical activity as one of six treatment goals [5]. However, its importance has only recently been recognized, and evidence demonstrating the effect of bronchodilators on physical activity is limited [30,31,32]. Furthermore, there are no randomized interventional studies focused on exercise capacity and physical activity in Japanese patients with COPD. Given that tiotropium is widely used as first-line monotherapy for maintenance management of COPD in Japan, the VESUTO® study has been planned to investigate the efficacy of tiotropium/olodaterol FDC compared to tiotropium monotherapy on exercise capacity (measured by 6MWD) and daily physical activity (measured by a 3-axis accelerometer).
This paper describes the study design of the VESUTO® study (NCT02629965), which is the first, randomized, 2-way crossover, interventional study (2 × 2 crossover study with 90 patients in each sequential group; in total, 180 patients/treatment group) in Japanese patients with COPD.
This study aims to assess the efficacy of tiotropium/olodaterol FDC compared with tiotropium monotherapy on IC as an index of lung hyperinflation in Japanese patients with COPD. In addition, the effect of these bronchodilators on exercise capacity and amount of daily physical activities will also be assessed.
Methods
Study Design
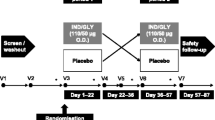
The VESUTO® study is a randomized, double-blind, active-controlled, 2-way crossover clinical study to assess the efficacy of once-daily administration of orally inhaled tiotropium/olodaterol FDC or tiotropium monotherapy on pulmonary function (lung hyperinflation), exercise capacity [6-min walk test (6MWT)], and physical activity measured by a 3-axis accelerometer in Japanese patients with COPD (NCT02629965). Patient follow-up in the study includes Visit 0 (informed consent), Visit 1 (screening), Visit 2 (randomization), Visit 3 to Visit 4 (assessment after crossover with 6-week treatment period), and Visit 5 (follow-up) (Fig. 1). This study protocol was finalized on October 30, 2015 as version 1.0. The study began in February 2016 in Japan.

Study design. 6MWT 6-min walk test, EOT end of treatment, FDC fixed-dose combination, IC inspiratory capacity, LABA long-acting β2 agonist, LAMA long-acting muscarinic antagonist, PFTs pulmonary function tests, V visit
The study will be carried out in compliance with the protocol, the principles laid down in the Declaration of Helsinki in accordance with the International Conference on Harmonisation (ICH) Guideline for Good Clinical Practice (GCP), relevant Boehringer Ingelheim Standard Operating Procedures, and the Japanese GCP regulations (Ministry of Health and Welfare Ordinance No. 28, March 27, 1997). The study was initiated only after all required legal documentation had been reviewed and approved by the respective Institutional Review Board and competent authority according to regulations in Japan. The detailed information of each Institutional Review Board is included in Supplementary Table S1. For inclusion, all patients must sign a written informed consent prior to participation in the study, which includes medication washout and restrictions. Standard medical care (prophylactic, diagnostic, and therapeutic procedures) remains the responsibility of the treating physician of the patient. Study data will be collected, analyzed, and published without personal identifiable information. The Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) checklist is provided in Supplementary Table S2.
Study Population and Setting
Men or women outpatients, aged ≥40 years with a diagnosis of COPD and stable airway obstruction, will be invited to participate in the study. Patients will be required to perform the 6MWT on several occasions during the study. Patients with any contraindication to exercise as stipulated in the European Respiratory Society (ERS) guidelines [33], and supported by the American Thoracic Society (ATS)/American College of Chest Physician guidelines will be excluded from participation [34]. The VESUTO® study has been planned to be conducted mainly at respiratory medicine centers of general clinics and hospitals located in major cities in Japan.
Inclusion and Exclusion Criteria
Patients will be required to have a diagnosis of COPD and stable airway obstruction with post-bronchodilator forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) <70%, post-bronchodilator FEV1 <80% of predicted normal, and GOLD grade 2–4 at Visit 1. Patients must be current or ex-smokers with a smoking history of >10 pack-years. Patients must also have a score of ≥1 on the modified Medical Research Council (mMRC) scale, 6MWD <400 m, and have a breathing discomfort on the modified Borg scale ≥4 measured just after the 6MWT at Visit 2. The criterion of the 6MWD <400 m is referred to in studies that evaluated the combination therapy of tiotropium and formoterol compared to tiotropium [22] and the ECLIPSE study which considered the relation of 6MWD with survival rate [23].
Main exclusion criteria are presence of a significant disease other than COPD; clinically relevant abnormal baseline hematology, blood chemistry, urinalysis, or creatinine more than twice the upper limit of normal; and current documented diagnosis of asthma. The main exclusion criteria are listed in Table 1.
A patient may be withdrawn from the study before completion if the patient withdraws consent or is no longer able to participate due to medical reasons [e.g., pregnancy, surgery, adverse events (AEs), or other diseases requiring discontinuation], or contraindications for exercise testing have occurred. Patients may also be withdrawn for administrative reasons (continuous protocol violations or persistent non-compliance of treatment administration).
Intervention
Eligible patients who consent to participate in the study will be randomized in a 1:1 ratio to two sequential groups at Visit 2 and will receive one of two treatments: tiotropium/olodaterol FDC 5/5 μg inhalation solution (2.5/2.5 μg per actuation) or tiotropium 5 μg inhalation solution (2.5 μg per actuation). These medications will be provided by Nippon Boehringer Ingelheim (Tokyo, Japan) and will be administered using the RESPIMAT® inhaler (Boehringer Ingelheim). The 6-week crossover treatment period does not include any washout period.
Procedure
Patients will be advised to self-administer the study medication at the hospital (study site) approximately at the same time at each clinic visit (Visit 2 ± 30 min and between 7:00 am and 10:00 am). Throughout the study, patients will self-administer the study medication at home, in the morning, in a seated position. Detailed written instructions and training for the use of the RESPIMAT® inhaler will be given to the patient at Visit 1 and repeated at Visits 2 and 3. The investigator or qualified study personnel will observe the inhalation procedure and will reinforce a correct inhalation technique. Salbutamol is permitted as rescue medication throughout the study.
Patients will complete a patient diary confirming that study medication was taken and record both the weather conditions and the number (puffs) of rescue medication use. The study medication compliance will be emphasized to be within 80–120% compliance rate.
For performing pulmonary function tests (PFTs), unified spirometers (Flowscreen; eResearch Technology GmbH; Estenfeld, Germany) will be provided by Boehringer Ingelheim at all investigational sites throughout the study. Spirometers and their use, including daily calibration, must meet ATS/ERS criteria [35]. The 6MWT will be performed according to the methodology described by ATS guidelines [34], and investigators and delegated qualified personnel are required to be trained for the 6MWT procedure before recruiting patients in order to control the variability of measurement by unifying the conduct of the procedure at each center.
At Visit 1, patients will be trained on the 6MWT before the PFT is performed. At Visit 2, the 6MWT for confirming eligibility and baseline measurement will be performed before administration of the first study medication. The 6MWT will be performed at the hospital at Visit 3 and Visit 4, starting 90 min (allowance +15 min) after administration of the study medication. During the 6MWT, oxygen saturation and heart rate will be monitored using a mobile pulse oximeter. Just after the end of the 6MWT, the intensity of breathing discomfort and leg discomfort will be assessed using a modified Borg Scale.
A physical activity monitor (HJA-750C; OMRON, Kyoto, Japan) will be used for assessment of physical activity. This is a 3-axis accelerometer that is used to measure the overall movement of a patient. Patients will wear the physical activity monitor every day for 2 weeks each prior to Visit 2 for baseline assessment, and prior to Visit 3 and Visit 4 for assessment of 6-week treatment. Patients will wear the physical activity monitor all day, except during bathing or water sport activities. Investigators will request patients to avoid extreme activities (e.g., extreme exercise) which are not a part of their normal daily routine in order to facilitate proper functioning of the equipment.
Rationale for Comparator, Dose, and Duration of the Treatments
Tiotropium/olodaterol is the FDC of tiotropium plus olodaterol, and we would like to know the additional effect to tiotropium, so we selected tiotropium monotherapy as comparator. The doses of both tiotropium/olodaterol FDC (5/5 µg) and tiotropium monotherapy (5 µg) were assessed in previous studies [10,11,12,13, 15, 16], and those doses were approved worldwide including Japan. The 6-week treatment period is the same period used in the previous studies assessing IC.
Endpoints
The primary endpoint is IC at rest, measured 60 min post-dose, and after 6-week treatment using FlowScreen from ERT®. The secondary endpoints include 6MWD (90 min post-dose), FVC (30 min post-dose), slow vital capacity (SVC) (60 min post-dose), and physical activity [average number of steps per day (steps/day); average daily duration (min) of ≥4 metabolic equivalents (METs); average daily duration (min) of ≥3 METs; average daily duration (min) of ≥2 METs; and average daily active strength (METs min) of ≥3 METs] after 6 weeks treatment.
Other efficacy endpoints are described in Table 2. Safety assessments will include heart rate, blood pressure, and oxygen saturation in conjunction during the 6MWT, and AEs will be monitored throughout the study.
Data which are planned to be collected at each visit are shown in Table 3.
Randomization and Blinding
Interactive Response Technology (IRT) will be used for randomization (ratio 1:1) to a treatment sequential group and for assignment of appropriate medications to eligible patients at Visit 2. Randomization will not be stratified. The IRT-assigned medication number, determined by a computer-generated random sequence, and the corresponding medication kit will be given to patients at Visit 2. Patients, investigators, and everyone involved in study conduct or analysis or with any other interest will remain blinded using this procedure.
Statistics
Study Populations
The randomized set will comprise all randomized patients, and the treated set (TS) will comprise patients who are documented to have taken any dose of study medication. The full analysis set (FAS), which will be used for primary endpoint analysis, will include TS patients with complete data at baseline Visit 2 and complete post-dose measurements of IC at rest. Assignment to the FAS will be done after implementation of any data handling rules, which set measurements to “missing”.
The per-protocol set (PPS) will contain a subset of patients for whom serious protocol deviations have not been identified. This subset will be defined under blinded review based on the magnitude and potential impact of any serious protocol violations.
Sample Size Calculation
For the primary endpoint, the difference in IC at rest after 6 weeks treatment between the two treatment groups is estimated to be 0.1 L. To detect a difference of 0.1 L with the standard deviation of 0.41 L (based on the previously conducted replicate studies [15]) in the IC at rest between the two treatments with 90% power at the 2-sided alpha of 0.05, 180 patients will be required. nQuery Advisor® + nTerim v.2.0 (MOT0-1, 1-sample t test) was used for the sample size calculation.
Primary and Secondary Endpoint Analyses
A mixed-effects model repeated measures (MMRM; SAS procedure MIXED) approach with treatment and period as categorical fixed effects, study baseline (Visit 2) as a covariate, and patient as a random effect will be used for primary endpoint analysis (IC at rest, measured at 60 min post-dose after 6 weeks treatment). Adjusted mean values, as well as treatment contrasts, will be presented with the 95% confidence intervals and values. If the number of patients in the PPS is <90% of the number of patients in the FAS, the primary analysis will also be performed on the PPS. These will be supportive analyses to assess the robustness of the primary analysis using the FAS. All secondary and further endpoints will be analyzed using a similar MMRM model. Descriptive statistics will be provided for each of the two treatment groups. Additionally, the analysis that the data generated during rainy weather is excluded will be performed as the sensitivity analysis to assess the impact of climate on the activity monitor [36].
Safety Analyses
Safety analyses will be conducted using the TS and will be mostly descriptive in nature. No hypothesis testing is planned prospectively. AEs will be coded using the Medical Dictionary for Regulatory Activities. The number of patients with AEs during the treatment period will be summarized, and the incidence will be compared across the treatment groups. Frequency, severity, seriousness, and causal relationship of AEs will be tabulated by system organ class and preferred term.
Data Acquisition and Handling of Missing Data
Data will be collected and captured through an electronic case report form. Missing data due to worsening of COPD symptoms leading to discontinuation of study medication will be imputed by the baseline data. In other cases, missing data will not be imputed.
Discussion
This multicenter, randomized, double-blind, active-controlled, 2-way crossover study aims to assess the efficacy of tiotropium/olodaterol FDC compared with tiotropium monotherapy on IC as an index of lung hyperinflation in Japanese patients with COPD. The effect of these bronchodilators on exercise capacity and amount of daily physical activities will also be assessed.
This is the first clinical study assessing a LAMA + LABA combination in Japanese patients with COPD to evaluate IC as a primary endpoint and exercise capacity assessed by 6MWD and physical activity measured by 3-axis accelerometer as secondary endpoints. The Japanese COPD guidelines specifically include improvement in exercise capacity and daily physical activity as one of the six treatment goals [5]; however, the evidence of exercise capacity or physical activity is limited in Japanese patients with COPD. Accordingly, evaluation of IC as an index of lung hyperinflation and its relationship with exercise capacity and physical activity in the VESUTO® study will provide additional evidence for COPD management.
The 6MWD is strongly associated with pulmonary hyperinflation [21], and the 6MWT will be performed to assess exercise capacity. Physical activity is potentially related to exercise capacity and is an independent prognostic factor in patients with COPD [36]. Decline in physical activity is also reported in association with poor prognosis and all-cause mortality in COPD [27, 28]. In the VESUTO® study, physical activity will be evaluated using a newly developed compact, 3-axis accelerometer. The measurements of physical activity with the 3-axis accelerometer are considered to be more objective than a questionnaire-based assessment.
Our VESUTO® study has several limitations such as inclusion of Japanese patients only, refined patients than in a clinical setting with a score of ≥1 on the mMRC scale, a 6MWD <400 m, and breathing discomfort as the modified Borg scale ≥4 measured just after the 6MWT. Additionally, since the treatment duration is only 6 weeks, assessment of the efficacy on exercise capacity and physical activity with longer treatment periods will provide valuable information on the long-term clinical benefit of bronchodilators.
Conclusions
In conclusion, the VESUTO® study should provide further evidence of tiotropium/olodaterol FDC compared to tiotropium in lung hyperinflation, exercise capacity, and physical activity for the comprehensive treatment of Japanese patients with COPD.
Trial Status
Ongoing.
References
National Institute for Health and Clinical Excellence (NICE). Chronic obstructive pulmonary disease in over-16s: diagnosis and management. NICE guidelines [CG101] London. 2010.
Fukuchi Y, Nishimura M, Ichinose M, et al. Chronic obstructive pulmonary disease (COPD) in Japan: the Nippon COPD epidemiology (NICE) study. Respirology. 2004;9:458–65.
Committee for the third edition of the COPD guidelines of the Japanese Respiratory Society. Guidelines for the diagnosis and treatment of COPD (chronic obstructive pulmonary disease) 3rd edition. Pocket guide. 2010.
From the Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD). 2016. http://goldcopd.org/. Accessed 13 August 2016.
Committee for the 4th edition of the COPD guidelines of the Japanese Respiratory Society. Guidelines for the diagnosis and treatment of COPD (chronic obstructive pulmonary disease) 4th edition. Pocket guide. 2013.
Cazzola M, Molimard M. The scientific rationale for combining long-acting beta2-agonists and muscarinic antagonists in COPD. Pulm Pharmacol Ther. 2010;23:257–67.
Tashkin DP, Ferguson GT. Combination bronchodilator therapy in the management of chronic obstructive pulmonary disease. Respir Res. 2013;14:49.
Matera MG, Rogliani P, Calzetta L, Cazzola M. Safety considerations with dual bronchodilator therapy in COPD: an update. Drug Saf. 2016;39:501–8.
Lahousse L, Verhamme KM, Stricker BH, Brusselle GG. Cardiac effects of current treatments of chronic obstructive pulmonary disease. Lancet Respir Med. 2016;4:149–64.
Buhl R, Maltais F, Abrahams R, et al. Tiotropium and olodaterol fixed-dose combination versus mono-components in COPD (GOLD 2-4). Eur Respir J. 2015;45:969–79.
Beeh KM, Westerman J, Kirsten AM, et al. The 24-h lung-function profile of once-daily tiotropium and olodaterol fixed-dose combination in chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2015;32:53–9.
Beeh KM, Derom E, Echave-Sustaeta J, et al. The lung function profile of once-daily tiotropium and olodaterol via Respimat® is superior to that of twice-daily salmeterol and fluticasone propionate via Accuhaler® (ENERGITO® study). Int J Chron Obstruct Pulm Dis. 2016;11:193–205.
Singh D, Ferguson GT, Bolitschek J, et al. Tiotropium + olodaterol shows clinically meaningful improvements in quality of life. Respir Med. 2015;109:1312–9.
Ichinose M. The efficacy and safety of combined tiotropium and olodaterol via the Respimat® inhaler in patients with COPD: results from the Japanese sub-population of the Tonado® studies. Int J Chron Obstruct Pulm Dis. 2016;11:2017–27.
O'Donnell D, Casaburi R, De Sousa D, et al. Effects of 6 weeks’ treatment with once-daily tiotropium and olodaterol fixed-dose combination on inspiratory capacity and exercise endurance in patients with COPD: the MORACTO™ studies. C22 I FEEL FINE NOW: NEW TREATMENTS FOR COPD. Am Thorac Soc. 2015;A3972.
Maltais F, Iturri JBG, Kirsten A, et al. Effects of 12 weeks of once-daily tiotropium and olodaterol fixed-dose combination on exercise endurance in patients with COPD. Eur Respir J. 2014;44:P283.
Oh YM, Bhome AB, Boonsawat W, et al. Characteristics of stable chronic obstructive pulmonary disease patients in the pulmonology clinics of seven Asian cities. Int J Chron Obstruct Pulm Dis. 2013;8:31–9.
Tan WC, Ng TP. COPD in Asia: where East meets West. Chest. 2008;133:517–27.
Cooper CB. The connection between chronic obstructive pulmonary disease symptoms and hyperinflation and its impact on exercise and function. Am J Med. 2006;119(10 Suppl 1):21–31.
Ferguson GT. Why does the lung hyperinflate? Proc Am Thorac Soc. 2006;3:176–9.
Cortopassi F, Celli B, Divo M, Pinto-Plata V. Longitudinal changes in handgrip strength, hyperinflation, and 6-minute walk distance in patients with COPD and a control group. Chest. 2015;148:986–94.
Jayaram L, Wong C, McAuley S, Rea H, Zeng I, O’Dochartaigh C. Combined therapy with tiotropium and formoterol in chronic obstructive pulmonary disease: effect on the 6-minute walk test. COPD. 2013;10:466–72.
Andrianopoulos V, Wouters EF, Pinto-Plata VM, et al. Prognostic value of variables derived from the six-minute walk test in patients with COPD: results from the ECLIPSE study. Respir Med. 2015;109:1138–46.
Garcia-Rio F, Lores V, Mediano O, et al. Daily physical activity in patients with chronic obstructive pulmonary disease is mainly associated with dynamic hyperinflation. Am J Respir Crit Care Med. 2009;180:506–12.
Troosters T, van der Molen T, Polkey M, et al. Improving physical activity in COPD: towards a new paradigm. Respir Res. 2013;14:115.
Minakata Y, Sugino A, Kanda M, et al. Reduced level of physical activity in Japanese patients with chronic obstructive pulmonary disease. Respir Investig. 2014;52:41–8.
Vaes AW, Garcia-Aymerich J, Marott JL, et al. Changes in physical activity and all-cause mortality in COPD. Eur Respir J. 2014;44:1199–209.
Waschki B, Kirsten A, Holz O, et al. Physical activity is the strongest predictor of all-cause mortality in patients with COPD: a prospective cohort study. Chest. 2011;140:331–42.
Ikeda N, Saito E, Kondo N, et al. What has made the population of Japan healthy? Lancet. 2011;378:1094–105.
Beeh KM, Watz H, Puente-Maestu L, et al. Aclidinium improves exercise endurance, dyspnea, lung hyperinflation, and physical activity in patients with COPD: a randomized, placebo-controlled, crossover trial. BMC Pulm Med. 2014;14:209.
Watz H, Krippner F, Kirsten A, Magnussen H, Vogelmeier C. Indacaterol improves lung hyperinflation and physical activity in patients with moderate chronic obstructive pulmonary disease—a randomized, multicenter, double-blind, placebo-controlled study. BMC Pulm Med. 2014;14:158.
Hataji O, Naito M, Ito K, Watanabe F, Gabazza EC, Taguchi O. Indacaterol improves daily physical activity in patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulm Dis. 2013;8:1–5.
Schünemann HJ, Woodhead M, Anzueto A, et al. A guide to guidelines for professional societies and other developers of recommendations: introduction to integrating and coordinating efforts in COPD guideline development. An official ATS/ERS workshop report. Proc Am Thor Soc. 2012;9:215–8.
American Thoracic Society/American College of Chest Physicians. ATS/ACCP Statement on cardiopulmonary exercise testing. Am J Respir Crit Care Med. 2003;167:211–77.
Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26:319–38.
Garcia-Rio F, Rojo B, Casitas R, et al. Prognostic value of the objective measurement of daily physical activity in patients with COPD. Chest. 2012;142:338–46.
Acknowledgements
We thank all patients and investigators of this VESUTO® study. This study was presented partially at the Asian Pacific Society of Respirology 2016 congress. The VESUTO® study was funded by Nippon Boehringer Ingelheim Co., Ltd., Tokyo, Japan. Nippon Boehringer Ingelheim contributed to the design of the study, and will be contributing to data collection, analysis, and interpretation of the study results. Nippon Boehringer Ingelheim has also funded the publication charges and open access fee for publication of this manuscript. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published. All authors contributed to the conception, design, or planning of the study. Masakazu Ichinose led the drafting of the manuscript, all authors contributed to critically revising the manuscript, and all authors read and approved the final manuscript. Editorial support, in the form of medical writing, assembling tables, and creating high-resolution images based on authors’ detailed directions, collating author comments, copyediting, fact checking, and referencing, was provided by Dr. Annirudha Chillar, MD, PhD, of Cactus Communications, and funded by Nippon Boehringer Ingelheim Co., Ltd, Tokyo, Japan. Scientific input, assistance with writing and editorial support was provided by Shuhei Nakamura, and editorial support, medical writing, and publication management was provided by Daisuke Kuroki, both of Nippon Boehringer Ingelheim Co., Ltd, Tokyo, Japan.
Disclosures
Masakazu Ichinose has received consulting fees from AstraZeneca, Nippon Boehringer Ingelheim, and Novartis Pharma KK, and has received honoraria from AstraZeneca, GSK, Nippon Boehringer Ingelheim, Kyorin, and Novartis Pharma. Yoshiaki Minakata and Takashi Motegi have nothing to disclose. Jun Ueki has received funding from Nippon Boehringer Ingelheim Co. Ltd., Otsuka Pharmaceutical Co. Ltd., and Teijin Pharma Ltd. Tetsuo Seki is an employee of Nippon Boehringer Ingelheim Co., Ltd. Tatsuhiko Anzai has received compensation from Nippon Boehringer Ingelheim Co., Ltd. for statistical analysis service. Ayako Takizawa is an employee of Nippon Boehringer Ingelheim Co., Ltd. Lars Grönke is an employee of Boehringer Ingelheim Pharma GmbH & Co. KG. Kazuto Hirata has received honorarium and travel grants from Nippon Boehringer Ingelheim Co., Ltd.
Compliance with Ethics Guidelines
The study will be carried out in compliance with the protocol, the principles laid down in the Declaration of Helsinki in accordance with the International Conference on Harmonisation (ICH) Guideline for Good Clinical Practice (GCP), relevant Boehringer Ingelheim Standard Operating Procedures, and the Japanese GCP regulations (Ministry of Health and Welfare Ordinance No. 28, March 27, 1997). The study will be initiated only after all required legal documentation has been reviewed and approved by the respective Institutional Review Board and competent authority according to regulations in Japan. The detailed information of each Institutional Review Board is included in Supplementary Table S1. For inclusion, all patients must sign a written informed consent prior to participation in the study, which includes medication washout and restrictions. Standard medical care (prophylactic, diagnostic, and therapeutic procedures) remains the responsibility of the treating physician of the patient. Study data will be collected, analyzed, and published without personal identifiable information. The Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) checklist is provided in Supplementary Table S2.
Data Availability
Datasets are currently unavailable due to the status of the trial.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/C028F0605AD94C93.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ichinose, M., Minakata, Y., Motegi, T. et al. Study Design of VESUTO®: Efficacy of Tiotropium/Olodaterol on Lung Hyperinflation, Exercise Capacity, and Physical Activity in Japanese Patients with Chronic Obstructive Pulmonary Disease. Adv Ther 34, 1622–1635 (2017). https://doi.org/10.1007/s12325-017-0554-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-017-0554-3