
Abstract
Introduction
Latanoprostene bunod is a novel nitric oxide (NO)-donating prostaglandin F2α receptor agonist in clinical development for the reduction of intraocular pressure (IOP) in patients with open-angle glaucoma or ocular hypertension. We evaluated the effect of latanoprostene bunod 0.024% instilled once daily (QD) on lowering IOP over a 24-h period in healthy Japanese subjects following 14 days of treatment.
Methods
This was a single-arm, single-center, open-label clinical study of 24 healthy Japanese male volunteers. A baseline IOP profile was established in both eyes in the sitting position at 8 PM, 10 PM, 12 AM, 2 AM, 4 AM, 8 AM, 10 AM, 12 PM, and 4 PM using a Goldmann applanation tonometer. Subjects subsequently instilled latanoprostene bunod 0.024% QD at 8 PM for 14 days in both eyes. The absolute and change from baseline in sitting IOP was assessed on day 14.
Results
The mean (SD) age of the subjects was 26.8 (6.3) years, and mean (SD) baseline IOP was 13.6 (1.3) mmHg in the study eye. Latanoprostene bunod 0.024% instilled QD for 14 days reduced IOP at all the evaluated time points (P < 0.001) with a mean (SD) 24-h reduction of 3.6 (0.8) mmHg or 27% from the baseline in the study eye. Peak and trough IOP lowering occurred at 8 AM and 8 PM (12 and 24 h following instillation) with a mean reduction of 4.2 (1.8) mmHg, or 30%, and 2.8 (2.2) mmHg, or 20%, respectively. Punctate keratitis and ocular hyperemia, both mild in severity, were the most common adverse events.
Conclusion
Latanoprostene bunod ophthalmic solution 0.024%, dosed QD for 14 days, significantly lowered mean IOP in healthy Japanese subjects during the entire 24-h period. Studies of latanoprostene bunod in patients diagnosed with normal tension glaucoma are warranted.
Trial Registration
Clinicaltrials.gov identifier NCT01895985.
Funding
Bausch & Lomb, Inc.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary open-angle glaucoma (POAG) is a chronic, progressive, optic neuropathy involving retinal ganglion cell death and optic nerve degeneration. Although a number of risk factors are associated with the development of POAG, including family history, age, race, intraocular pressure (IOP), and use of topical or systemic corticosteroids, the reduction of IOP is the only proven method of treatment for the disease [1, 2]. Normal tension glaucoma (NTG) is defined as POAG with an IOP of ≤21 mmHg, and accounts for a significant proportion of POAG cases, particularly in the Asian population [3, 4]. The Collaborative Normal Tension Glaucoma Study demonstrated that, despite being within the normal range, IOP is a factor in the optic nerve damage observed in NTG and that therapy aimed at lowering IOP is beneficial in patients at risk of progression [5, 6]. Further studies have suggested that reduced blood flow, leading to reduced ocular perfusion pressure, and/or diurnal IOP fluctuation and nocturnal IOP spikes contribute to disease progression in NTG [7–10].
Latanoprostene bunod (LBN; Bausch & Lomb, Inc.) is a single entity nitric-oxide (NO)-donating prostaglandin F2α receptor agonist in clinical development for the reduction of intraocular pressure (IOP) in patients with open-angle glaucoma or ocular hypertension (OHT). On topical ocular administration, LBN is rapidly metabolized by esterases to latanoprost acid, a prostaglandin analog, and butanediol mononitrate (BDMN), the nitric oxide NO-donating moiety. Nitric oxide is subsequently released from BDMN in conjunction with 1,4-butanediol, an inactive metabolite. Latanoprost acid, the active moiety present in Xalatan® (latanoprost ophthalmic solution 0.005%), lowers IOP through a mechanism that mainly involves long-term remodeling of the extracellular matrices in the ciliary body, thereby increasing uveoscleral (nonconventional) aqueous humor outflow [11–15]. Nitric oxide is an endogenous signaling mediator which has been shown to lower IOP in clinical and animal models of OHT [16–21] and has been implicated in IOP homeostasis in POAG patients [22, 23]. Nitric oxide is reported to mediate IOP lowering in animal and ex vivo tissue models through an increase in conventional (trabecular meshwork and Schlemm’s canal) aqueous outflow [18, 19, 24–29]. This appears to be mediated though activation of a signaling cascade involving elevation of cyclic guanosine 3′, 5′-monophosphate levels and subsequent relaxation of the tissues of the conventional outflow pathway [28–31].
Nonclinical and clinical studies have demonstrated the contribution of both active moieties of LBN (latanoprost acid and NO) to its robust IOP-lowering efficacy, thereby suggesting a dual mechanism of action for LBN [32–34]. Krauss et al. demonstrated that LBN lowered IOP with greater efficacy than equimolar concentrations of latanoprost in multiple animal models of elevated IOP [33]. More recently, Cavet et al. reported that LBN had a significantly greater relaxation effect on endothelin-1 contracted human trabecular meshwork cells than that observed with latanoprost [32]. Weinreb et al. further showed that LBN administered once daily (QD) was effective in lowering IOP in subjects with OAG or OHT in a dose-dependent manner with LBN 0.024% dose providing statistically significantly greater reductions in mean diurnal IOP than latanoprost 0.005% QD (Xalatan) [34].
The objective of the present clinical study, designated the KRONUS study (Clinicaltrials.gov identifier: NCT01895985), was to evaluate the effect of LBN 0.024% dosed QD in reducing IOP measured over a 24-h monitoring period in healthy, male Japanese subjects. Determination of time points for peak and trough IOP lowering effects were of interest for the design of future pivotal efficacy studies of LBN in Japanese patients with POAG or OHT.
Methods
Study Design
This single-arm, open-label study was conducted at a single clinical site located in Japan. The three study visits occurred in a clinical setting, and all study procedures were conducted by certified investigators and/or technicians. All study-related documents, including the protocol, informed consent form, and subject recruitment materials were approved by the Kitasato University Kitasato Shirogane Institutional Review Board (Shirogane, Minato-ku, Tokyo, Japan). This study was conducted in accordance with current International Conference on Harmonization (ICH), Good Clinical Practice (GCP) guidelines, Ministerial Ordinance on GCP for Drugs (JAPAN GCP), and the Declaration of Helsinki 7th revision (2013).
Subjects
Subjects who participated in this study were recruited from a pool of healthy volunteers. All subjects were required to be male, at least 20 years of age, and be able to self-administer eye drops. A normal ophthalmic history and corrected decimal visual acuity (VA) of 0.5 or better in both eyes were also required for study enrollment. Subjects were not allowed to use non-prescription (including vitamins or dietary supplements) or prescription drugs 28 days prior to study drug administration or during the study treatment period (14 days). Additionally, subjects were excluded from the study if they had a history of severe ocular trauma or incisional surgery, a history of ocular laser surgery (90 days), or had any intraocular inflammation or infection within 90 days prior to screening.
Study Treatments and Assessments
All subjects instilled LBN ophthalmic solution 0.024% (manufactured by Bausch & Lomb Inc, Tampa, FL) QD for 14 days at approximately 8 PM, starting on the evening of day 1 and ending on the evening of day 14, and completed three study visits (Fig. 1). Visit 1, the screening visit, occurred from 1 to 28 days prior to Visit 2, the baseline visit. At the screening visit, subjects were evaluated against the inclusion and exclusion criteria and, if found to be suitable for enrollment, vital signs, corrected decimal VA, a slit-lamp examination, IOP measurements, and ophthalmoscopy assessments were conducted. A urine sample for drug testing and a blood sample for serology testing were also collected. At screening and at all study visits, IOP was determined in both eyes using Goldmann applanation tonometry in the sitting position. Adverse events were collected at all study visits.

Schematic of the clinical study design. Three study visits were required for all subjects to complete the study. Measurements of intraocular pressure (IOP) were recorded from both eyes for each subject at Visit 2 and Visit 3 at nine time points (8 PM, 10 PM, 12 AM, 2 AM, 4 AM, 8 AM, 10 AM, 12 PM, and 4 PM). Latanoprostene bunod 0.024% was provided to all subjects following the 4 PM IOP assessments at Visit 2, with instructions to instill the study drug QD at 8 PM
Visit 2, the baseline visit, occurred on days 0–1. Intraocular pressure measurements were taken at 8 PM and 10 PM, marking the beginning of the 24-h baseline IOP measurements. Additional IOP measurements were taken at 12 AM (marking the beginning of day 1), 2 AM, 4 AM, 8 AM, 10 AM, 12 PM, and 4 PM. Study drug was dispensed to subjects following the 4 PM assessment. Prior to discharge, subjects were instructed to instill the first drop of study drug into the conjunctival cul de sac of each eye at approximately 8 PM on day 1, which was the start of the treatment period, and were instructed to record their daily dosing on a diary card. Both eyes were to be dosed QD at 8 PM for the duration of the study.
Visit 3 was the exit visit, occurring on days 14–15. Subjects returned to the clinic within 1 h prior to instillation of the final dose at 8 PM in the clinic on day 14. Intraocular pressure measurements were taken at approximately 8 PM and 10 PM, marking the beginning of the 24-h IOP measurements while on treatment. Additional IOP measurements were taken at 12 AM (marking the beginning of day 15), 2 AM, 4 AM, 8 AM, 10 AM, 12 PM, and 4 PM. Additional safety assessments at Visit 3 included vital signs (resting blood pressure and pulse), VA, and slit-lamp examination.
Statistical Methods
The intent-to-treat (ITT) population included all subjects who received at least one dose of study drug and had at least one post-baseline IOP assessment. The safety population included all subjects who received at least one dose of study drug. The per-protocol (PP) population included all subjects in the ITT population who remained in the study through Visit 3 (day 14/15) and who did not have major protocol deviations. It was assumed that a sample size of 20 evaluable subjects would provide sufficient data to evaluate the IOP profile over 24 h using descriptive statistics.
The absolute and change from baseline (CFB) in IOP at day 14/15 were summarized using descriptive statistics (n, mean, SD) for each assessment time point. In addition, a paired t test was performed on the CFB (ITT and PP populations). For the purpose of evaluating the primary efficacy endpoint (CFB in IOP at each measurement time point), the right eye was considered the study eye. A P value less than or equal to 0.05 was the threshold for determining statistical significance. Further, baseline and post-baseline mean 24-h IOP was calculated by taking the average of the nine IOP values (8 PM, 10 PM, 12 AM, 2 AM, 4 AM, 8 AM, 10 AM, 12 PM, 4 PM) for each subject’s eyes (study eye and fellow eye, separately) and determining the mean (SD) of those. All statistical analyses were conducted using SAS version 9.1 (SAS Institute, Inc., Cary NC).
Adverse events (AEs) were summarized using discrete summaries at the subject and event level by system organ class and preferred term. Ophthalmoscopy and slit-lamp examination measurements were summarized using descriptive statistics for discrete variables. Visual acuity was summarized using descriptive statistics for continuous variables.
Dosing compliance was assessed based on returned subject diaries using the following calculation: \( {\text{Percent compliance = }}\left[ { 1 0 0 { } \,\times {\text{ number of instillations received}}} \right]{ / }\left[ {{\text{date of diary collection {-} date of first dose + 1}}} \right] . \)
Results
Subject Disposition and Demographics
A total of 45 subjects were screened and 24 subjects were enrolled in the study. All enrolled subjects completed the assessments required for the three study visits. There were no major protocol violations; hence the ITT and PP populations were the same. The mean (SD) age of the study subjects was 26.8 (6.3) years (range 20–39 years). All subjects were male, as specified by the study inclusion criteria. Mean (SD) 24-h baseline IOP was 13.6 (1.3) mmHg in the study eye and 13.9 (1.3) mmHg in the fellow eye.
All subjects were 81–120% compliant with dosing instructions based on the information recorded in the subject diaries. Twenty-two subjects administered all 14 daily doses and two subjects administered 13 out of 14 daily doses.
Efficacy Results
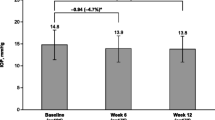
The baseline (Visit 2) and post-treatment (Visit 3) mean IOP values for the study eye and treated fellow eye for subjects in the ITT population are presented by assessment time point in Fig. 2a, b, respectively. The mean CFB in IOP values for the study eye and treated fellow eye is presented by assessment time point in Fig. 3. Treatment with LBN 0.024% QD for 14 days significantly reduced IOP at all time points evaluated (P < 0.001). Mean (SD) 24-h IOP post-treatment was 10.0 (1.0) mmHg in the study eye and 10.3 (1.0) mmHg in the treated fellow eye; the mean (SD) 24-h reduction in IOP was 3.6 (0.8) mmHg or 27% from baseline in the study eye, and 3.5 (0.9) mmHg or 25% from baseline in the treated fellow eye. The peak and trough IOP lowering effect occurred at 8 AM and 8 PM (12 and 24 h following instillation) with a mean (SD) reduction from baseline in the study eye of 4.2 (1.8) mmHg, or 30% at peak, and 2.8 (2.2) mmHg, or 20% at trough, respectively. Peak and trough IOP lowering was also observed at 8 AM and 8 PM for the treated fellow eye, with mean (SD) reductions from baseline of 4.5 (1.8) mmHg, or 31%, and 2.8 (2.3) mmHg, or 18%, respectively.

Mean intraocular pressure (IOP) over 24 h at baseline and after 2 weeks of treatment with latanoprostene bunod, 0.024% in the study eye (a), and the treated fellow eye (b)

Change from baseline in mean intraocular pressure (IOP) over 24 h after 2 weeks of treatment with latanoprostene bunod, 0.024%. The mean change from baseline (CFB) ± standard error (mmHg) for the study eye and the treated fellow eye are depicted. Statistically significant differences were observed at all measured time points over the 24-h monitoring period for the CFB in mean IOP assessments in both eyes (P < 0.0001; paired t test)
Safety Results
No serious adverse events were reported during the course of the study. A total of 55 ocular and one non-ocular treatment-emergent adverse events (TEAEs) were reported. One subject reported the occurrence of a non-ocular TEAE during the study (mild upper respiratory inflammation) that was considered unrelated to study treatment. The 55 ocular TEAEs occurred in 22 subjects (28 associated with the study eye; 27 associated with the treated fellow eye), and were all determined to be mild; the majority of ocular TEAEs were judged to be at least possibly or probably related to the study drug treatment [study eye 96.4% (27/28), fellow eye 96.3% (26/27)]. Punctate keratitis and conjunctival hyperemia were the most commonly reported adverse events (Table 1).
There were no meaningful changes in vital signs or VA from screening to Visit 3. Mean (SD) systolic/diastolic blood pressure was 111.1 (10.7)/66.9 (9.7) at baseline and 103.9 (10.2)/61.3 (9.8) post-treatment; mean heart rate was 64.9 (9.7) beats per minute (bpm) at baseline and 65.8 (11.4) bpm post-treatment. The mean (SD) corrected decimal VA at baseline was 1.06 (0.35) and 1.08 (0.35) for the study eyes and fellow eyes, respectively. Post-treatment mean (SD) corrected decimal VA was 1.04 (0.33) and 1.07 (0.35) for the study eyes and fellow eyes, respectively.
Ophthalmoscopy and biomicroscopy findings were normal with few exceptions: mild papilla in the palpebral conjunctiva in one subject (both eyes, Visit 3), mild superficial punctate keratitis in four subjects (both eyes in three subjects; n = 1 Visit 1, n = 2 Visits 1 and 2, n = 1 Visit 3), and mild cataract in one subject at Visit 1 and Visit 3.
Conjunctival hyperemia was graded at screening and at Visit 3 on a 4-point scale (none, mild, moderate, severe) with the aid of photographic standards. At baseline, 79.2% (19/24) of subjects had no indication of conjunctival hyperemia, 16.7% (4/24) of subjects had mild conjunctival hyperemia, and 4.2% (1/24) of subjects had moderate conjunctival hyperemia. Post-treatment, 45.8% (11/24) of subjects had no indication of conjunctival hyperemia and 54.2% (13/24) of subjects had mild conjunctival hyperemia.
Discussion
The purpose of this single-center, open-label study (KRONUS) was to evaluate the effect of LBN 0.024%, a single entity monotherapy, administered once daily for 2 weeks on IOP over a 24-h monitoring period as compared to baseline in healthy Japanese subjects. LBN significantly lowered mean IOP at all measured time points over the 24-h period to an overall mean of 10 mmHg after 2 weeks of treatment. The effect of LBN included dampening of the diurnal rise in IOP (8 AM) that was observed at baseline in both the study and fellow eyes. Given the importance of IOP fluctuations in the progression of glaucoma [35, 36], in particular in NTG [37, 38], the sustained reduction of IOP over 24 h is of note. LBN 0.024% was safe and well-tolerated by the study population; all adverse events recorded during the study were mild in severity, and there was no meaningful effect on vital signs or visual acuity. Changes in conjunctival hyperemia were not unexpected as subjects were administered anesthetic drops every 2 h prior to IOP measures. Both anesthetics and prostaglandin analogs may increase levels of hyperemia.
The Collaborative Normal-Tension Glaucoma study demonstrated that IOP was a risk factor for NTG and suggested that IOP lowering may help reduce the progression of NTG [5, 6]. Other studies have shown that progression of visual field damage is slowed in NTG patients after trabeculectomy which lowered IOP to <11 mmHg [39, 40]. In the current study of healthy subjects, overall, a 27% reduction in IOP was noted over 24-h monitoring in subjects with a baseline mean IOP of 13.6 mmHg in the study eye. These results are promising and warrant the study of LBN 0.024% in subjects with a diagnosis of NTG. Further, studies have shown that IOP in non-glaucomatous eyes of Japanese subjects is lower as compared to IOP in eyes of subjects with white or black ethnicity [41, 42]. In this context, it is noteworthy that in the current study instillation of LBN resulted in a robust reduction in IOP, despite the subjects’ low baseline IOP.
The IOP reduction observed with LBN in the current study compares favorably to that observed with other IOP-lowering monotherapies in either healthy subjects or patients with NTG. Latanoprost 0.005% administered QD in the evening was reported to reduce IOP (mean diurnal or a single daily measurement) between 15 and 23% in studies varying from 7 to 24 days duration conducted in healthy subjects [43–46]. Similarly, QD administration of latanoprost 0.005% reduced diurnal or 24-h mean IOP by 14–24% in NTG patients after 3–12 weeks of treatment [47–54]. Additional studies in NTG patients have shown a 20% reduction in diurnal IOP after timolol administered twice daily for 3 weeks [47], a 16% reduction in 24-h IOP after bimatoprost administered QD for 8 weeks [54], and a 24% reduction in IOP (mean of two daytime measurements) after bimatoprost administration QD for 2 weeks [55]. Studies with travoprost QD in NTG patients have shown an 11% reduction in mean IOP over 24 h after 4 weeks [56] and a 19.4% decrease in mean IOP measured within 2 h after drug administration in the evening after 12 weeks of treatment [57].
Conclusion
The results of the present study demonstrate the ability of LBN 0.024% to provide a robust IOP lowering over 24 h after 2 weeks of dosing in healthy subjects. Taken with previously reported IOP lowering data with LBN 0.024% in patients with POAG or OHT [34], the results of the present study suggest the potential of LBN 0.024% to reduce IOP in glaucoma patients not only with elevated IOP but also with IOP in the normal range. Studies of LBN 0.024% in patients diagnosed with NTG are warranted.
References
Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311(18):1901–11.
Weinreb RN, Khaw PT. Primary open-angle glaucoma. Lancet. 2004;363(9422):1711–20.
Iwase A, Suzuki Y, Araie M, et al. The prevalence of primary open-angle glaucoma in Japanese: the Tajimi Study. Ophthalmology. 2004;111(9):1641–8.
Sommer A, Tielsch JM, Katz J, et al. Relationship between intraocular pressure and primary open angle glaucoma among white and black Americans: the Baltimore eye survey. Arch Ophthalmol. 1991;109(8):1090–5.
Anderson DR. Collaborative normal tension glaucoma study. Curr Opin Ophthalmol. 2003;14(2):86–90.
Collaborative Normal-Tension Glaucoma Study Group. Comparison of glaucomatous progression between untreated patients with normal-tension glaucoma and patients with therapeutically reduced intraocular pressures. Am J Ophthalmol. 1998;126(4):487–97.
Deokule S, Weinreb RN. Relationships among systemic blood pressure, intraocular pressure, and open-angle glaucoma. Can J Ophthalmol. 2008;43(3):302–7.
Hayreh SS, Zimmerman MB, Podhajsky P, Alward WL. Nocturnal arterial hypotension and its role in optic nerve head and ocular ischemic disorders. Am J Ophthalmol. 1994;117(5):603–24.
Meyer JH, Brandi-Dohrn J, Funk J. Twenty four hour blood pressure monitoring in normal tension glaucoma. Br J Ophthalmol. 1996;80(10):864–7.
Quaranta L, Katsanos A, Russo A, Riva I. 24-hour intraocular pressure and ocular perfusion pressure in glaucoma. Surv Ophthalmol. 2013;58(1):26–41.
Gabelt BT, Kaufman PL. Prostaglandin F2 alpha increases uveoscleral outflow in the cynomolgus monkey. Exp Eye Res. 1989;49(3):389–402.
Lindsey JD, Kashiwagi K, Kashiwagi F, Weinreb RN. Prostaglandins alter extracellular matrix adjacent to human ciliary muscle cells in vitro. Invest Ophthalmol Vis Sci. 1997;38(11):2214–23.
Lutjen-Drecoll E, Tamm E. Morphological study of the anterior segment of cynomolgus monkey eyes following treatment with prostaglandin F2 alpha. Exp Eye Res. 1988;47(5):761–9.
Nilsson SF, Samuelsson M, Bill A, Stjernschantz J. Increased uveoscleral outflow as a possible mechanism of ocular hypotension caused by prostaglandin F 2α-1-isopropylester in the cynomolgus monkey. Exp Eye Res. 1989;48(5):707–16.
Richter M, Krauss AH, Woodward DF, Lutjen-Drecoll E. Morphological changes in the anterior eye segment after long-term treatment with different receptor selective prostaglandin agonists and a prostamide. Invest Ophthalmol Vis Sci. 2003;44(10):4419–26.
Chuman H, Chuman T, Nao-i N, Sawada A. The effect of l-arginine on intraocular pressure in the human eye. Curr Eye Res. 2000;20(6):511–6.
Kotikoski H, Alajuuma P, Moilanen E, et al. Comparison of nitric oxide donors in lowering intraocular pressure in rabbits: role of cyclic GMP. J Ocul Pharmacol Ther. 2002;18(1):11–23.
Nathanson JA. Nitrovasodilators as a new class of ocular hypotensive agents. J Pharmacol Exp Ther. 1992;260(3):956–65.
Schuman JS, Erickson K, Nathanson JA. Nitrovasodilator effects on intraocular pressure and outflow facility in monkeys. Exp Eye Res. 1994;58(1):99–105.
Stamer WD, Lei Y, Boussommier-Calleja A, Overby DR, Ethier CR. eNOS, a pressure-dependent regulator of intraocular pressure. Invest Ophthalmol Vis Sci. 2011;52(13):9438–44.
Wizemann AJ, Wizemann V. Organic nitrate therapy in glaucoma. Am J Ophthalmol. 1980;90(1):106–9.
Doganay S, Evereklioglu C, Turkoz Y, Er H. Decreased nitric oxide production in primary open-angle glaucoma. Eur J Ophthalmol. 2002;12(1):44–8.
Galassi F, Renieri G, Sodi A, Ucci F, Vannozzi L, Masini E. Nitric oxide proxies and ocular perfusion pressure in primary open angle glaucoma. Br J Ophthalmol. 2004;88(6):757–60.
Heyne GW, Kiland JA, Kaufman PL, Gabelt BT. Effect of nitric oxide on anterior segment physiology in monkeys. Invest Ophthalmol Vis Sci. 2013;54(7):5103–10.
Kee C, Kaufman PL, Gabelt BT. Effect of 8-Br cGMP on aqueous humor dynamics in monkeys. Invest Ophthalmol Vis Sci. 1994;35(6):2769–73.
Kotikoski H, Vapaatalo H, Oksala O. Nitric oxide and cyclic GMP enhance aqueous humor outflow facility in rabbits. Curr Eye Res. 2003;26(2):119–23.
Wiederholt M, Sturm A, Lepple-Wienhues A. Relaxation of trabecular meshwork and ciliary muscle by release of nitric oxide. Invest Ophthalmol Vis Sci. 1994;35(5):2515–20.
Ellis DZ, Dismuke WM, Chokshi BM. Characterization of soluble guanylate cyclase in NO-induced increases in aqueous humor outflow facility and in the trabecular meshwork. Invest Ophthalmol Vis Sci. 2009;50(4):1808–13.
Schneemann A, Dijkstra BG, van den Berg TJ, Kamphuis W, Hoyng PF. Nitric oxide/guanylate cyclase pathways and flow in anterior segment perfusion. Graefes Arch Clin Exp Ophthalmol. 2002;240(11):936–41.
Cavet ME, Vittitow JL, Impagnatiello F, Ongini E, Bastia E. Nitric oxide (NO): an emerging target for the treatment of glaucoma. Invest Ophthalmol Vis Sci. 2014;55(8):5005–15.
Dismuke WM, Mbadugha CC, Ellis DZ. NO-induced regulation of human trabecular meshwork cell volume and aqueous humor outflow facility involve the BKCa ion channel. Am J Physiol Cell Physiol. 2008;294(6):C1378–86.
Cavet ME, Vollmer TR, Harrington KL, VanDerMeid K, Richardson ME. Regulation of endothelin-1-induced trabecular meshwork cell contractility by latanoprostene bunod. Invest Ophthalmol Vis Sci. 2015;56(6):4108–16.
Krauss AH, Impagnatiello F, Toris CB, et al. Ocular hypotensive activity of BOL-303259-X, a nitric oxide donating prostaglandin F2α agonist, in preclinical models. Exp Eye Res. 2011;93(3):250–5.
Weinreb RN, Ong T, Scassellati Sforzolini B, Vittitow JL, Singh K, Kaufman PL. A randomised, controlled comparison of latanoprostene bunod and latanoprost 0.005% in the treatment of ocular hypertension and open angle glaucoma: the VOYAGER study. Br J Ophthalmol. 2014;99(6):738–45.
Asrani S, Zeimer R, Wilensky J, Gieser D, Vitale S, Lindenmuth K. Large diurnal fluctuations in intraocular pressure are an independent risk factor in patients with glaucoma. J Glaucoma. 2000;9(2):134–42.
Bergea B, Bodin L, Svedbergh B. Impact of intraocular pressure regulation on visual fields in open-angle glaucoma. Ophthalmology. 1999;106(5):997–1004.
Caprioli J, Coleman AL. Intraocular pressure fluctuation a risk factor for visual field progression at low intraocular pressures in the advanced glaucoma intervention study. Ophthalmology. 2008;115(7):1123–1129e3.
Sakata R, Aihara M, Murata H, et al. Intraocular pressure change over a habitual 24-hour period after changing posture or drinking water and related factors in normal tension glaucoma. Invest Ophthalmol Vis Sci. 2013;54(8):5313–20.
Koseki N, Araie M, Shirato S, Yamamoto S. Effect of trabeculectomy on visual field performance in central 30 degrees field in progressive normal-tension glaucoma. Ophthalmology. 1997;104(2):197–201.
Shigeeda T, Tomidokoro A, Araie M, Koseki N, Yamamoto S. Long-term follow-up of visual field progression after trabeculectomy in progressive normal-tension glaucoma. Ophthalmology. 2002;109(4):766–70.
Sommer A, Tielsch JM, Katz J, et al. Relationship between intraocular pressure and primary open-angle glaucoma among white and black Americans. The Baltimore eye survey. Arch Ophthalmol. 1991;109:1090–5.
Iwase A, Suzuki Y, Araie M, et al. The prevalence of primary open-angle glaucoma in Japanese: the Tajimi Study. Ophthalmology. 2004;111(9):1641–8.
Darhad UNM, Fujioka M, Tatsumi Y, Nagai-Kusuhara A, Maeda H, Negi A. Intraocular pressure lowering effect of once daily versus once weekly latanoprost instillation in the same normal individuals. Kobe J Med Sci. 2007;53(6):297–304.
Kawaguchi I, Higashide T, Ohkubo S, Kawaguchi C, Sugiyama K. Comparison of efficacy of four prostaglandin analogues by bilateral treatment in healthy subjects. Jpn J Ophthalmol. 2012;56(4):346–53.
Lim KS, Nau CB, O’Byrne MM, et al. Mechanism of action of bimatoprost, latanoprost, and travoprost in healthy subjects. A crossover study. Ophthalmology. 2008;115(5):790–795 e4.
Takahashi M, Higashide T, Sakurai M, Sugiyama K. Discrepancy of the intraocular pressure response between fellow eyes in one-eye trials versus bilateral treatment: verification with normal subjects. J Glaucoma. 2008;17(3):169–74.
Drance SM, Crichton A, Mills RP. Comparison of the effect of latanoprost 0.005% and timolol 0.5% on the calculated ocular perfusion pressure in patients with normal-tension glaucoma. Am J Ophthalmol. 1998;125(5):585–92.
Ishibashi S, Hirose N, Tawara A, Kubota T. Effect of latanoprost on the diurnal variations in the intraocular and ocular perfusion pressure in normal tension glaucoma. J Glaucoma. 2006;15(5):354–7.
Kondo N, Sawada A, Yamamoto T, Taniguchi T. Correlation between individual differences in intraocular pressure reduction and outflow facility due to latanoprost in normal-tension glaucoma patients. Jpn J Ophthalmol. 2006;50(1):20–4.
Liu CJ, Ko YC, Cheng CY, et al. Changes in intraocular pressure and ocular perfusion pressure after latanoprost 0.005% or brimonidine tartrate 0.2% in normal-tension glaucoma patients. Ophthalmology. 2002;109(12):2241–7.
McKibbin M, Menage MJ. The effect of once-daily latanoprost on intraocular pressure and pulsatile ocular blood flow in normal tension glaucoma. Eye Lond. 1999;13(Pt 1):31–4.
Nakamoto K, Yasuda N. Effect of concomitant use of latanoprost and brinzolamide on 24-hour variation of IOP in normal-tension glaucoma. J Glaucoma. 2007;16(4):352–7.
Rulo AH, Greve EL, Geijssen HC, Hoyng PF. Reduction of intraocular pressure with treatment of latanoprost once daily in patients with normal-pressure glaucoma. Ophthalmology. 1996;103(8):1276–82.
Quaranta L, Pizzolante T, Riva I, Haidich AB, Konstas AG, Stewart WC. Twenty-four-hour intraocular pressure and blood pressure levels with bimatoprost versus latanoprost in patients with normal-tension glaucoma. Br J Ophthalmol. 2008;92(9):1227–31.
Tsumura T, Yoshikawa K, Suzumura H, et al. Bimatoprost ophthalmic solution 0.03% lowered intraocular pressure of normal-tension glaucoma with minimal adverse events. Clin Ophthalmol. 2012;6:1547–52.
Seibold LK, Kahook MY. The diurnal and nocturnal effects of travoprost in normal-tension glaucoma. Clin Ophthalmol. 2014;8:2189–93.
Mizoue S, Nakano T, Fuse N, Iwase A, Matsumoto S, Yoshikawa K. Travoprost with sofZia(R) preservative system lowered intraocular pressure of Japanese normal tension glaucoma with minimal side effects. Clin Ophthalmol. 2014;8:347–54.
Acknowledgments
Sponsorship, article processing charges, and the open access charge for this study were funded by Bausch & Lomb, Inc. Medical writing support was provided by Kurt Brubaker, BS, Bridge Over Brook (Medford, OR) and funded by Bausch & Lomb, Inc. Editorial assistance was provided by Sandra Westra, PharmD of Churchill Communications (Maplewood, NJ), also funded by Bausch & Lomb, Inc. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Presentations
These data were previously reported at the Association for Research in Vision and Ophthalmology (ARVO) meeting, Orlando, FL, May 4–8, 2014.
Grant Support
None.
Disclosures
Makoto Araie is a paid consultant for Bausch & Lomb, Inc.; Baldo Scassellati Sforzolini was an employee of Bausch & Lomb, Inc. during the conduct of this study (currently employed by Allergan, Inc); Jason Vittitow is an employee of Bausch & Lomb, Inc.; Robert N. Weinreb has served as a paid consultant for Bausch & Lomb, Alcon, and Allergan, and has received grants from Genentech, Heidelberg Engineering, Quark, Nidek, National Eye Institute, Aerie, Topcon, and Aquesys.
Compliance with ethics guidelines
All study-related documents, including the protocol, informed consent form, and subject recruitment materials were approved by the Kitasato University Kitasato Shirogane Institutional Review Board (Shirogane, Minato-ku, Tokyo, Japan). This study was conducted in accordance with current International Conference on Harmonization (ICH), Good Clinical Practice (GCP) guidelines, Ministerial Ordinance on GCP for Drugs (JAPAN GCP), and the Declaration of Helsinki 7th revision (2013).
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Araie, M., Sforzolini, B.S., Vittitow, J. et al. Evaluation of the Effect of Latanoprostene Bunod Ophthalmic Solution, 0.024% in Lowering Intraocular Pressure over 24 h in Healthy Japanese Subjects. Adv Ther 32, 1128–1139 (2015). https://doi.org/10.1007/s12325-015-0260-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-015-0260-y