
Abstract
Introduction
Atopic dermatitis (AD) has been related to a deficiency of delta-6-desaturase, an enzyme responsible for the conversion of linoleic acid to gamma-linolenic acid (GLA). Evening primrose oil (EPO) contains high amounts of GLA. Therefore, this study investigated whether EPO supplementation results in an increase in plasma GLA and its metabolite dihomo-gamma-linolenic acid (DGLA) correlating with clinical improvement of AD, assessed by the SCORing Atopic Dermatitis (SCORAD) index.
Methods
The open study included 21 patients with AD. EPO (4–6 g) was administered daily for 12 weeks. Before treatment, and 4 and 12 weeks after initiation of EPO supplementation, objective SCORAD was assessed and plasma concentrations of GLA and DGLA were determined by gas chromatography.
Results
A significant increase in plasma GLA and DGLA levels and a decrease in the objective SCORAD were observed 4 and 12 weeks after initiation of EPO treatment. In the per-protocol population (n = 14), a significant inverse correlation between the changes in plasma GLA levels and SCORAD was found (P = 0.008).
Conclusion
The clinical disease activity under EPO treatment correlates with the individual increase in plasma GLA levels. Thus, the results of this pilot study indicate that an increase in plasma GLA might be used as predictive parameter for responsiveness of AD to EPO therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atopic dermatitis (AD) is a chronic inflammatory skin disorder characterized by eczematous lesions, skin dryness, and severe itch [1]. AD affects 15–30% of children and 2–10% of adults [1]. The etiopathology of AD is multifactorial and includes a genetic predisposition towards disrupted skin barrier function and allergic inflammation as well as environmental factors [2]. Defects of the skin barrier (e.g., due to mutations of the filaggrin gene) lead to an increased irritability followed by inflammatory processes [3].
At least in a subset of patients with AD, a malfunction of delta-6-desaturase seems to play a pathogenetic role [4]. This enzyme is responsible for the conversion of linoleic acid (LA) to gamma-linolenic acid (GLA), which is further metabolized to dihomo-gamma-linolenic acid (DGLA). DGLA is the precursor of prostaglandin E1 (PGE1) or 15-hydroxyeicosatrienoic acid (15-HETrE) [5]. As a result of a reduced enzyme activity of delta-6-desaturase, high levels of LA and low levels of GLA have been observed in AD patients [6]. Accordingly, less DGLA and its metabolites PGE1 and 15-HETrE known to have anti-inflammatory capacity are produced [5].
Supplementation with GLA, a component of evening primrose oil (EPO), is assumed to lead to increased concentrations of GLA and DGLA in blood, thereby supporting anti-inflammatory responses [7]. Indeed, GLA supplementation studies in humans and rodents have shown that the synthesis of anti-inflammatory PGE1, but not pro-inflammatory PGE2, was selectively elevated [8].
The EPO preparation used in this study contained high amounts of GLA (80 mg/1,000 mg EPO) with sufficient bioavailability after oral intake [9]. The clinical efficacy of EPO supplementation in AD has been shown in numerous studies [10–12]. Nevertheless, the use of EPO has been judged controversially [10, 13, 14]; moreover, a measurable biomarker for the possible efficacy of EPO has not yet been described.
This study aimed to investigate whether a clinical response to EPO supplementation in AD patients correlates with an increase in plasma GLA and DGLA levels. It should be defined whether GLA and DGLA would, therefore, represent possible biomarkers. With that, patients responding to EPO supplementation with clinical improvement of AD could be identified by determining GLA and/or DGLA plasma levels.
Methods
Study Design
The study was designed as a prospective, explorative, multi-center, open, non-controlled pilot trial at five centers in Switzerland. It was conducted according to ICH guidelines for Good Clinical Practice. All procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2000 and 2008. Informed oral and written consent was obtained from all patients and/or their legal representatives for being included in the study. The study protocol was approved by the cantonal ethics committees and the regulatory agency. The study was registered at ClinicalTrials.gov (Identifier: NCT00878670).
Patients and Clinical Assessments
Patients between 2 and 45 years old were included into the study if they had AD according to the criteria of Hanifin and Rajka [15] with predominant rough and fissured skin as well as pruritus for at least 2 months. Women of childbearing potential needed to use a highly effective method of contraception during the entire duration of the study. Pregnant or lactating women were excluded.
The main exclusion factors were the presence of chronic dermatosis such as seborrheic dermatitis, contact dermatitis, nummular eczema, psoriasis, ichthyosis, an immuno-deficiency or any immunological disorder, scabies, cutaneous fungal infection, HIV-associated skin disorders, malignant diseases, T cell-Lymphoma, Letterer-Siwe-disease, progressive systemic diseases, serious internal diseases (e.g., serious de-compensated diseases of the heart, liver, and/or kidneys, or diabetes mellitus) or hypersensitivity towards one of the ingredients in the investigational product. Study participants who had been taking part in another study or had taken an investigational product during the last 4 weeks before the start of treatment were not allowed to take part in the study.
The study participants were allowed to continue to use any medication which they had been taking prior to the study at the same dose, unless the medication could be discontinued or was listed as prohibited medication. Prohibited medication prior to study start and during the study were as follows. Thirty days prior to study start, physical or psychological therapy, anti-inflammatory medication to treat AD, and immuno-modulating medication were not allowed. Fourteen days prior to study start, non-steroidal anti-rheumatic drugs, systemic use of glucocorticosteroids, tranquillizers or antiemetic agents from the phenothiazine group were forbidden and 7 days prior to study start alpha- or beta-blockers, clonidine, alpha-sympathomimetic medication, azelastine, levocabastine, or antidepressants were not allowed to be taken by the patient.
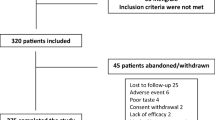
The objective SCORing Atopic Dermatitis (SCORAD) index [16] and plasma GLA and DGLA levels were assessed before treatment (baseline) and 4 and 12 weeks after treatment initiation. As safety parameters, the occurrence of adverse events (AEs), hematological and clinical-chemical laboratory tests, and vital signs were assessed. A total of 36 patients were screened and 13 screening failures occurred; forbidden medication (pimecrolimus, antidepressants; n = 2), holidays (n = 2), refusal of blood sampling (n = 1), no interest to participate (n = 1), not possible to participate (n = 1), and no reason specified (n = 6).
Study Medication
The study medication (EPOGAM® 1000, Zeller Medical AG, Romanshorn, Switzerland) contained 932–1,073 mg EPO (Oenothera seminis oleum), corresponding to 80 mg GLA per capsule. The dosages were two capsules twice daily for patients aged 2–12 years and three capsules twice daily for patients over 12 years. Treatment duration was 12 weeks. Treatment compliance was defined as intake of at least 75% of the trial medication. Rescue medication including prednicarbate cream 0.25% (Prednitop®, Abbott AG, Baar, Switzerland) and hydroxyzine syrup 2 mg/ml (Atarax®; UCB Pharma, Bulle, Switzerland) was disposed to coup eczema exacerbations and pruritus, respectively.
Blood Sample Analysis
The DGLA and GLA measurements were performed at AZ Biopharm GmbH (Berlin, Germany). To stabilize fatty acids, butylated hydroxytoluene was added and blood was stored at −20 °C. As published elsewhere [17], absolute levels of GLA and DGLA in human plasma were determined applying a gas chromatographic method with mass selective detection. Additionally, safety laboratory parameters [routine parameters of blood count and blood chemistry as well as Immunoglobulin E (IgE) levels] were measured locally at each investigator’s site laboratory.
Statistics
To compare SCORAD and fatty acid levels at baseline, week 4 and week 12, data were tested by non-parametric Wilcoxon Signed Rank Test for paired samples for the intention-to-treat (ITT) population. The relationship between the clinical response (percentage reduction of objective SCORAD from baseline to last visit) and the increase in GLA and DGLA in plasma from baseline to last visit was assessed by linear regression models in the per-protocol (PP) population including only patients with acceptable treatment compliance. The possible effect of the co-medication on the clinical improvement (relative change of objective SCORAD) was assessed using a non-parametric group comparison (users versus non-users) by means of Kruskal–Wallis test. The statistical analysis was performed with the software SPSS version 19.0 (SPSS Inc., Chicago, IL, USA).
Results
Patients
A total of 36 patients were screened, of whom 23 were included in the study (safety population; 14 females). The mean age was 26.3 years (range 3–58 years). The age groups were as follows: under 12 years (n = 6), between 12 and 18 years (n = 2), and above 18 years (n = 15). Eighty-seven percent of the patients were Caucasian. Two patients were lost to follow-up, resulting in 21 patients in the ITT population. From the ITT population, seven patients had to be excluded because of poor treatment compliance (<75% of study medication intake). Thus, the PP population included 14 patients.
In total, 14 patients of the safety population (n = 23) had allergic concomitant diseases such as asthma bronchiale, allergic rhinitis, or rhinoconjunctivitis. No significant difference in the SCORAD index between patients of the PP population (n = 14) with allergic concomitant diseases (n = 8) and patients without (n = 6) could be found in a sub-analysis (data not shown).
Objective SCORAD Significantly Decreased
The clinical efficacy of EPO was assessed in the ITT population by recording the objective SCORAD at baseline, 4 weeks and 12 weeks after initiation of EPO therapy. At baseline, 42.9% of the patients had mild symptoms (objective SCORAD <25) and 57.1% had moderate symptoms (objective SCORAD 25–50), according to Oranje et al. [16]. After the 12-week treatment period, 93.2% of the patients had mild symptoms, whereas only 6.8% had moderate symptoms. Accordingly, the mean objective SCORAD showed a time-dependent reduction with a statistically significant decrease after 4 and 12 weeks, respectively, compared to baseline (Fig. 1; Supplemental Table 1 in the Electronic Supplementary Material). With specific focus on SCORAD parameters, a significant decrease in both the extent and total intensity of AD after EPO treatment was noticed (Table 1). In addition, the intensity of individual AD symptoms except for weeping/crusting was significantly reduced. Supplementation of EPO had no effect on total IgE levels (data not shown).

Objective SCORAD (%, mean ± SEM) in the intention-to-treat population (n = 21) assessed at baseline and 4 and 12 weeks after initiation of EPO treatment (Wilcoxon Signed Rank Test). EPO evening primrose oil, SCORAD SCORing Atopic Dermatitis
Total SCORAD Significantly Decreased
In line with the results obtained for the objective SCORAD, the clinical efficacy of EPO could also be confirmed for the total SCORAD. The mean total SCORAD showed a time-dependent reduction with a statistically significant decrease after 4 (P = 0.019) and 12 weeks (P = 0.001), respectively. Results of all individual SCORAD parameters are illustrated in Table 1.
Increase in Plasma Fatty Acid Levels Inversely Correlated with SCORAD
In the ITT population, the plasma levels of both GLA and DGLA showed a significant increase over the study period (Fig. 2; Supplemental Table 1 in the Electronic Supplementary Material). This increase was most evident from baseline to week 4, with no relevant further increase to week 12. For the PP population, it could be shown that the reduction of the objective SCORAD was inversely correlated with the increase in plasma GLA (Fig. 3). In the ITT population, the correlation of the objective SCORAD and plasma GLA was not significant (data not shown) For DGLA, no significant correlation could be demonstrated (P = 0.36; data not shown).

Plasma GLA (a) and DGLA (b) levels (mean ± SEM) in the intention-to-treat population (n = 21) at baseline and 4 and 12 weeks after initiation of EPO treatment (Wilcoxon Signed Rank Test). DGLA dihomo-gamma-linolenic acid, EPO evening primrose oil, GLA gamma-linolenic acid

Correlation analysis on the per-protocol population (n = 14) between the change in objective SCORAD (%) and change of plasma GLA (%) from baseline versus last visit. GLA gamma-linolenic acid, SCORAD SCORing Atopic Dermatitis
Usage of Rescue Medication did not Affect Study Outcome
While EPO represents a long-term basic therapy for AD, the usage of concomitant treatment options reflects the usual practice. Patients were allowed to use the provided rescue medication (prednicarbate cream and hydroxyzine syrup), for example, in cases of severe itching. The influence of the co-medication was evaluated in the ITT population (n = 21). During the study, rescue medication was used by the majority of patients (71.4%). A total of nine patients used hydroxyzine (syrup, 10–50 mg orally). Twelve patients used prednicarbate (cream, 0.25% topically) as rescue medication. Six of these patients used both rescue medications. The treatment duration and dosage regimens varied quite differently in both rescue medications (Table 2). However, the patient groups relying on rescue medication did not show any significant difference regarding the improvement of the objective SCORAD compared to the group without usage of concomitant anti-pruritic medication (P = 0.20 and P = 0.89, respectively).
Safety
During the study, 26 AEs were recorded in 13 patients. No serious AEs occurred. Only five AEs were assessed to be related to the study drug (tiredness, impaired concentration, diarrhea, abdominal cramps, and pruritus). No clinically relevant abnormalities of safety laboratory parameters or vital signs were noticed.
Discussion
This study showed that supplementation with EPO had significant effects on clinical improvement of clinical signs in patients with AD in association with an increase in plasma GLA and DGLA levels. This is in line with previous studies, revealing the clinical effect of EPO after 4–8 weeks of treatment [10]. The present study showed further improvement of AD symptoms upon 12 weeks, supporting the idea that EPO should be applied as long-term basic therapy.
Low peripheral blood levels of GLA and DGLA have been reported in patients with AD [18]. Supplementation with EPO containing relevant amounts of GLA resulted in a significant increase in GLA and DGLA plasma levels in our AD patient group. Since GLA and DGLA can be metabolized to 15-HETrE and PGE1 in the skin, a deficit in GLA might result in a reduced production of these anti-inflammatory metabolites [5]. Thus, low GLA levels might contribute to inflammation of allergic diseases. Accordingly, GLA levels inversely correlated with total serum IgE levels [18].
It has been shown that in children with AD receiving GLA supplementation have a lower SCORAD compared to the placebo group [19]. The increase in plasma GLA concentration was significantly inversely correlated with AD severity [19]. Moreover, a placebo-controlled study investigating the effect of borage oil containing at least 23% GLA showed a clinical improvement of AD, in particular, in those patients in whom an increase in erythrocyte DGLA levels was found [20]. In agreement with these reports, a significant reduction of clinical signs in a group of pediatric and adult AD patients treated with EPO as assessed by objective SCORAD was observed. Moreover, the increase in GLA plasma levels upon supplementation with EPO was associated with a reduction of the objective SCORAD. This effect was observed as early as 4 weeks after initiating EPO therapy.
Remarkably, the significance of these preliminary results could be obtained despite the small sample size. The difference in plasma GLA levels might be an appropriate marker to identify responders. Thus, AD patients benefitting from EPO could be selected 4 weeks after initiating supplementation and be given advice to continue EPO supplementation. Patients without an increase in plasma GLA after 4 weeks although receiving EPO supplementation might most likely be regarded as non-responders and recommended to stop therapy.
The limitation of this study was the open, non-randomized, non-placebo-controlled design. An additional linear regression analysis was applied to further strengthen the correlation between plasma GLA levels and clinical response. Since the number of patients was rather small and the group was heterogeneous, further studies are required to evaluate the relation between GLA levels and AD severity in larger pediatric and adult cohorts. Such an approach might set a cut-off level clearly distinguishing between responders and non-responders.
Conclusion
Supplementation therapy with EPO resulted in a significant increase in plasma GLA and DGLA levels. The increase in plasma GLA was associated with an improvement of clinical signs in patients with AD. Thus, the difference in GLA levels upon EPO therapy after 4 weeks could serve as biomarker to identify responders among AD patients.
References
Bieber T. Atopic dermatitis. N Engl J Med. 2008;358(14):1483–94.
Wuthrich B, Cozzio A, Roll A, Senti G, Kundig T, Schmid-Grendelmeier P. Atopic eczema: genetics or environment? Ann Agric Environ Med. 2007;14(2):195–201.
Simon D, Kernland Lang K. Atopic dermatitis: from new pathogenic insights toward a barrier-restoring and anti-inflammatory therapy. Curr Opin Pediatr. 2011;23(6):647–52.
Horrobin DF. Fatty acid metabolism in health and disease: the role of delta-6-desaturase. Am J Clin Nutr. 1993;57(5 Suppl):732S–6S (discussion 6S-7S).
Ziboh VA, Miller CC, Cho Y. Metabolism of polyunsaturated fatty acids by skin epidermal enzymes: generation of antiinflammatory and antiproliferative metabolites. Am J Clin Nutr. 2000;71(1 Suppl):361S–6S.
Yen CH, Dai YS, Yang YH, Wang LC, Lee JH, Chiang BL. Linoleic acid metabolite levels and transepidermal water loss in children with atopic dermatitis. Ann Allergy Asthma Immunol. 2008;100(1):66–73.
Horrobin DF. Essential fatty acid metabolism and its modification in atopic eczema. Am J Clin Nutr. 2000;71(1 Suppl):367S–72S.
Fan YY, Chapkin RS. Importance of dietary gamma-linolenic acid in human health and nutrition. J Nutr. 1998;128(9):1411–4.
Martens-Lobenhoffer J, Meyer FP. Pharmacokinetic data of gamma-linolenic acid in healthy volunteers after the administration of evening primrose oil (Epogam). Int J Clin Pharmacol Ther. 1998;36(7):363–6.
Morse NL, Clough PM. A meta-analysis of randomized, placebo-controlled clinical trials of Efamol evening primrose oil in atopic eczema. Where do we go from here in light of more recent discoveries? Curr Pharm Biotechnol. 2006;7(6):503–24.
Morse PF, Horrobin DF, Manku MS, et al. Meta-analysis of placebo-controlled studies of the efficacy of Epogam in the treatment of atopic eczema. Relationship between plasma essential fatty acid changes and clinical response. Br J Dermatol. 1989;121(1):75–90.
van Gool CJ, Zeegers MP, Thijs C. Oral essential fatty acid supplementation in atopic dermatitis-a meta-analysis of placebo-controlled trials. Br J Dermatol. 2004;150(4):728–40.
Williams HC. Evening primrose oil for atopic dermatitis. BMJ. 2003;327(7428):1358–9.
Williams HC, Grindlay DJ. What’s new in atopic eczema? An analysis of the clinical significance of systematic reviews on atopic eczema published in 2006 and 2007. Clin Exp Dermatol. 2008;33(6):685–8.
Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Arch Dermatol. 1980;113:663–70.
Oranje AP, Glazenburg EJ, Wolkerstorfer A, de Waard-van der Spek FB. Practical issues on interpretation of scoring atopic dermatitis: the SCORAD index, objective SCORAD and the three-item severity score. Br J Dermatol. 2007;157(4):645–8.
Schulte E, Weber K. Fast generation of fatty acid methly esters from lipids using trimethyl sulfonium hydroxide or sodium methylate [German]. Eur J Lipid Sci Technol. 1989;5:181–3.
Focke M, Sesztak-Greinecker G, Brannath W, Gotz M, Jarisch R, Hemmer W. Plasma levels of polyunsaturated fatty acids in children with atopic dermatitis and in atopic and nonatopic controls. Wien Klin Wochenschr. 2005;117(13–14):485–91.
van Gool CJ, Thijs C, Henquet CJ, et al. Gamma-linolenic acid supplementation for prophylaxis of atopic dermatitis–a randomized controlled trial in infants at high familial risk. Am J Clin Nutr. 2003;77(4):943–51.
Henz BM, Jablonska S, van de Kerkhof PC, et al. Double-blind, multicentre analysis of the efficacy of borage oil in patients with atopic eczema. Br J Dermatol. 1999;140(4):685–8.
Acknowledgments
Sponsorship and article processing charges for this study was funded by Max Zeller Söhne AG, Romanshorn, Switzerland. EPO (EPOGAM® 1000) was provided by Max Zeller Söhne AG. Prof. P. Schmid-Grendelmeier is the guarantor for this article, and takes responsibility for the integrity of the work as a whole.
Conflict of interest
C. Zimmermann is an employee of Max Zeller Söhne AG. C. Zahner is an employee of Max Zeller Söhne AG. J. Drewe is an employee of Max Zeller Söhne AG. L. Hess has received consulting fees from Max Zeller Söhne AG. Peter Schmid-Grendelmeier, D. Simon, P. Eng, S. Borelli, R. Kägi, G. Ferrari, S. Lautenschlager and B. Wüthrich have no conflict of interest to declare.
Compliance with ethics guidelines
This study was conducted according to ICH guidelines for Good Clinical Practice. All procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2000 and 2008. Informed oral and written consent was obtained from all patients and/or their legal representatives for being included in the study. The study protocol was approved by the cantonal ethics committees and the regulatory agency.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
ClinicalTrials.gov #NCT00878670.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Simon, D., Eng, P.A., Borelli, S. et al. Gamma-Linolenic Acid Levels Correlate with Clinical Efficacy of Evening Primrose Oil in Patients with Atopic Dermatitis. Adv Ther 31, 180–188 (2014). https://doi.org/10.1007/s12325-014-0093-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-014-0093-0