
Abstract
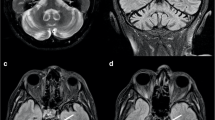
Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS) is now increasingly identified from all countries over the world, possibly rendering it one of the most common autosomal recessive ataxias. Here, we selected patients harboring SACS variants, the causative gene for ARSACS, in a large cohort of 137 patients with early-onset ataxia recruited from May 2019 to May 2021 and were referred to the ataxia clinic. Genetic studies were performed for 111 out of 137 patients (81%) which led to a diagnostic rate of 72.9% (81 out of 111 cases). Ten patients with the molecular diagnosis of ARSACS were identified. We investigated the phenotypic and imaging spectra of all confirmed patients with ARSACS. We also estimated the frequency of ARSACS in this cohort and described their clinical and genetic findings including seven novel variants as well as novel neuroimaging findings. While the classic clinical triad of ARSACS is progressive cerebellar ataxia, spasticity, and sensorimotor polyneuropathy, it is not a constant feature in all patients. Sensorimotor axonal-demyelinating neuropathy was detected in all of our patients, but spasticity and extensor plantar reflex were absent in 50% (5/10). In all patients, brain magnetic resonance imaging (MRI) showed symmetric linear hypointensities in the pons (pontine stripes) and anterior superior cerebellar atrophy as well as a hyperintense rim around the thalami (thalamic rim). Although infratentorial arachnoid cyst has been reported in ARSACS earlier, we report anterior temporal arachnoid cyst in two patients for the first time, indicating that arachnoid cyst may be an associated imaging feature of ARSACS. We also extended molecular spectrum of ARSACS by presenting 8 pathogenic and one variant of unknown significance (VUS) sequence variants, which 7 of them have not been reported previously. MetaDome server confirmed that the identified VUS variant was in the intolerant regions of sacsin protein encoded by SACS.



Similar content being viewed by others

Data Availability
Human variants and pertinent phenotypes have been reported to ClinVar (Submission IDs: SUB9677044, SUB9677045, SUB9677050, SUB9677685, SUB9677710, SUB9677755, SUB9677767, SUB9677775).
References
Vermeer S, Meijer RP, Pijl BJ, Timmermans J, Cruysberg JR, Bos MM, et al. ARSACS in the Dutch population: a frequent cause of early-onset cerebellar ataxia. Neurogenetics. 2008;9(3):207–14.
Synofzik M, Soehn AS, Gburek-Augustat J, Schicks J, Karle KN, Schüle R, et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis. 2013;8(1):1–13.
Synofzik M, Nemeth AH. Recessive ataxias. Handb Clin Neurol. 2018;155:73–89.
Xiromerisiou G, Dadouli K, Marogianni C, Provatas A, Ntellas P, Rikos D, et al. A novel homozygous SACS mutation identified by whole exome sequencing-genotype phenotype correlations of all published cases. J Mol Neurosci. 2020;70(1):131–41.
Habibzadeh P, Tabatabaei Z, Inaloo S, Nashatizadeh MM, Synofzik M, Ostovan VR, et al. Case report: expanding the genetic and phenotypic spectrum of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Front Genet. 2020;11:585136.
Artero Castro A, Machuca C, Rodriguez Jimenez FJ, Jendelova P, Erceg S. Short review: investigating ARSACS: models for understanding cerebellar degeneration. Neuropathol Appl Neurobiol. 2019;45(6):531–7.
Parfitt DA, Michael GJ, Vermeulen EG, Prodromou NV, Webb TR, Gallo J-M, et al. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum Mol Genet. 2009;18(9):1556–65.
Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, et al. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc Natl Acad Sci USA. 2009;106(16):6706–11.
Breckpot J, Takiyama Y, Thienpont B, Van Vooren S, Vermeesch JR, Ortibus E, et al. A novel genomic disorder: a deletion of the SACS gene leading to spastic ataxia of Charlevoix-Saguenay. Eur J Hum Genet. 2008;16(9):1050–4.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine. 2015;17(5):405–23.
Mohammadi P, Heidari M, Ashrafi MR, Mahdieh N, Garshasbi MG. A novel homozygous missense variant in the NAXE gene in an Iranian family with progressive encephalopathy with brain edema and leukoencephalopathy. Acta Neurol Belg. 2021. https://doi.org/10.1007/s13760-021-01717-y.
Mohammadi P, Salehi Siavashani E, Mohammadi MF, Bahramy A, Almadani N, Garshasbi MG. Whole-exome sequencing identified first homozygous frameshift variant in the COLEC10 gene in an Iranian patient causing 3MC syndrome type 3. Mol Genet Genomic Med. 2021;9:e1834.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90.
Abuín JM, Pichel JC, Pena TF, Amigo J. BigBWA: approaching the Burrows-Wheeler aligner to Big Data technologies. Bioinformatics. 2015;31(24):4003–5.
Fattahi Z, Beheshtian M, Mohseni M, Poustchi H, Sellars E, Nezhadi SH, et al. Iranome: a catalogue of genomic variations in the Iranian population. Hum Mutat. 2019;40(11):1968–84.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31(10):1536–43.
Parkinson MH, Bartmann AP, Clayton LM, Nethisinghe S, Pfundt R, Chapple JP, et al. Optical coherence tomography in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Brain. 2018;141(4):989–99.
Hosseini-Chavoshi M, Abbasi-Shavazi MJ, Bittles AH. Consanguineous marriage, reproductive behaviour and postnatal mortality in contemporary Iran. Hum Hered. 2014;77(1–4):16–25.
Engert JC, Doré C, Mercier J, Ge B, Bétard C, Rioux JD, et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): high-resolution physical and transcript map of the candidate region in chromosome region 13q11. Genomics. 1999;62(2):156–64.
Pilliod J, Moutton S, Lavie J, Maurat E, Hubert C, Bellance N, et al. New practical definitions for the diagnosis of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Ann Neurol. 2015;78(6):871–86.
Oguz K, Haliloglu G, Temucin C, Gocmen R, Has A, Doerschner K, et al. Assessment of whole-brain white matter by DTI in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Am J Neuroradiol. 2013;34(10):1952–7.
Chan JL, Tan AL, Ng LP, Low DC, Tew SW, Low SY. Paediatric arachnoid cysts: surgical outcomes from a Singapore children’s hospital. J Clin Neurosci. 2021;85:122–31.
Dlaka D, Raguž M, Muller D, Romić D, Almahariq F, Dlaka J, et al. Intraparenchymal supratentorial arachnoid cyst: a case report. Egypt J Neurosurg. 2019;34(1):1–6.
Rezende Filho FM, Parkinson MH, Pedroso JL, Poh R, Faber I, Lourenço CM, et al. Clinical, ophthalmological, imaging and genetic features in Brazilian patients with ARSACS. Parkinsonism Relate Disord. 2019;62:148–55.
Chang Y-F, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74.
Hammer MB, Eleuch-Fayache G, Gibbs JR, Arepalli SK, Chong SB, Sassi C, et al. Exome sequencing: an efficient diagnostic tool for complex neurodegenerative disorders. Eur J neurol. 2013;20(3):486–92.
Acknowledgements
The authors thank all participant families and their patients in this research. The authors are especially thankful to Ali Mohebi and Nahid Vafaee from the Growth and Development Research Center, Tehran University of Medical Sciences, for their support in data gathering. The authors thank the Hertie Institute for Clinical Brain Research, Tübingen, Germany, as the international collaborative party of the study.
Funding
This project was supported by the Deutsche Forschungsgemeinschaft (DFG) (German Research Foundation) No. 441409627, as part of the PROSPAX consortium under the frame of EJP RD, the European Joint Programme on Rare Diseases, under the EJP RD COFUND-EJP N° 825575. This study was granted by NIMAD under the proposal No. 971846.
Author information
Authors and Affiliations
Contributions
MRA and MH designed and supervised the study. ZR, ART, PM, and NM had a major contribution in drafting the manuscript. ZR, MH, ART, SH, MR, MR, RAM, RSB, DF, AZD, and AR interpreted clinical data. ART and MS contributed in the final scientific revision. NM, PM, and SS contributed in genetic data analyses. MGA interpreted the electrodiagnostic data. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
This study was approved by the ethics committee of the National Institute for Medical Research Development of Iran (Ethics ID: IR.NIMAD.REC.1397.508) and has thus been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Consent to Participate
Informed consent was obtained from all individual participants enrolled in the study.
Consent for Publication
Participant families agreed on anonymous publication of patients’ clinical information and their relevant data.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mahmoud Reza Ashrafi and Pouria Mohammadi have an equal contribution as the first authors.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ashrafi, M.R., Mohammadi, P., Tavasoli, A.R. et al. Clinical and Molecular Findings of Autosomal Recessive Spastic Ataxia of Charlevoix Saguenay: an Iranian Case Series Expanding the Genetic and Neuroimaging Spectra. Cerebellum 22, 640–650 (2023). https://doi.org/10.1007/s12311-022-01430-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-022-01430-3