
Abstract
Spinocerebellar ataxia type 19 (SCA19), allelic with spinocerebellar ataxia type 22 (SCA22), is a rare syndrome caused by mutations in the KCND3 gene which encodes the potassium channel Kv4.3. Only 18 SCA19/22 families and sporadic cases of different ethnic backgrounds have been previously reported. As in other SCAs, the SCA19/22 phenotype is variable and usually consists of adult-onset slowly progressive ataxia and cognitive impairment; myoclonus and seizures; mild Parkinsonism occurs in some cases. Here we describe a Swedish SCA19/22 family spanning five generations and harboring the T377M mutation in KCND3. For the first time for this disease, 18F-fluorodeoxyglucose PET was assessed revealing widespread brain hypometabolism. In addition, we identified white matter abnormalities and found unusual features for SCA19/22 including early age of onset and fast rate of progression in the late course of disease in the oldest patient of this family.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinocerebellar ataxias (SCA) are a heterogeneous and growing group of autosomal dominant diseases enumerated in chronological order from SCA1 to SCA43 according to the current classification in use [1]. Pathological CAG expansions in different genes cause SCA1, SCA2, SCA3, SCA6, and SCA7 which represent up to 60% of all SCA cases; the remaining SCA subtypes are rare and in most cases associated with conventional mutations [2, 3]. Spinocerebellar ataxia types 19 (SCA19) and 22 (SCA22) are rare allelic channelopathies; linkage to chromosome region 1p21-q21 was reported first in two large Dutch and Chinese families [4, 5]; and the mutations in KCND3, which encodes the Shal-related voltage-gated potassium channel Kv4.3, associated with ataxia were discovered in 2012 by two independent research groups [6,7,8]. Previously, variants and mutations in KCND3 were found to be associated with sudden unexpected death syndrome (SUDS) and later with Brugada syndrome-9 (BRGDA9) [9,10,11]. So far, only one ataxia patient harboring the mutation L450P in KCND3 has been diagnosed with Brugada syndrome A. [10, 11]. Only 18 SCA19/22 families and sporadic cases of diverse ethnicities have been described so far [6, 7, 10,11,12,13,14,15]. In addition, two variants of unclear significance (VUS) associated with ataxia have been identified in a recent screening study [12]. The SCA19/SCA22 phenotype consists of adult-onset and slowly progressive cerebellar ataxia in most cases, frequent cognitive impairment and variable degree of myoclonus, polyneuropathy, and seizures. Mild Parkinsonism has been reported recently in two unrelated French SCA19 families [14]. The Thr377Met (T377M) mutation in KCND3 has been described only once in a Japanese patient affected by pure cerebellar ataxia [7]. The aim of this study was to perform a comprehensive characterization of four affected members of a Swedish family spanning five generations; all affected family members harbor the T377M mutation in KCND3. This characterization includes clinical and cognitive evaluations, structural brain imaging and, for the first time for this disease, brain 18F-fluorodeoxyglucose PET ([18F] FDG PET).
Materials and Methods
Patients
Four patients (III:1, III:2, IV:1, and V:1) from the same family of Swedish origin were recruited and underwent standard clinical investigation, psychometric testing, neuroimaging studies, electroneurography (ENeG) and genetic analyses. This study was carried out in accordance with the recommendations of the ethics committee in Stockholm and the radiation protection organization at the Karolinska University Hospital (Etikprövningsnämnden dnr 2016/2538-32) with written informed consent from all patients. All patients gave written informed consent in accordance with the Declaration of Helsinki. Disease status in three affected shown in the pedigree was assigned by history (Fig. 1). The following scales were used during the clinical evaluation: Assessment and Rating of Ataxia (SARA) and Inventory of non-ataxia Symptoms (INAS); neuropsychological testing of the index case III:1 (2015) and patient V:1 (2016) was carried out with the following batteries: (1) brief cognitive status: Montreal Cognitive Assessment (MoCA) and Mini Mental State Examination (MMSE); (2) general intellectual ability (IQ): Raven’s progressive matrices; (3) evaluation of verbal episodic memory: Rey Auditory Verbal Learning Test (RAVLT); (4) visuospatial episodic memory: Rey-Osterrieth Complex Figure Test (ROCFT); (5) working memory: digit span of the Wechsler Adult Intelligence Scale (WAIS-III); (6) spatial/visual construction: ROCFT, Copy and Block Design/WAIS; (7) verbal concept formation: similarities in WAIS-III; (8) word fluency: Controlled Oral Word Association Test (FAS/COWAT); (9) picture naming: Boston Naming Test (BNT); (10) information processing speed: Symbol Digit Modalities Test (SDMT); (11) Executive function: Trail Making Test, B (TMT); and (12) motor speed: finger-tapping test (FT), dominant and non-dominant hand. Z scores, computed on the basis of reference values from test manuals and handbooks, were used to compare results from various tests [16]; [17], a z score ≤ − 1.5 SD is compatible with a significant cognitive deficit.

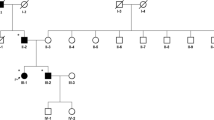
Pedigree of a Swedish SCA19/22 family. Disease status in generations I and II was asigned by history. All four tested patients (III:1, III:2, IV:1 and V:1) harbor the T377M mutation in KCND3.Variable degrees of axial ataxia, polyneuropathy, executive deficits, cerebellar atrophy and white matter abnormalities were identified
Imaging
Standard neuroimaging with 3T MRI scanners were performed in all four patients, an experienced neuroradiologist did a qualitative assessment. Brain metabolism was evaluated with [18 F] FDG PET in three patients at the Karolinska University Hospital in Huddinge. Imaging acquisition after intravenous injection of 18F-FDG (2 MBq/kg) was performed in a Biograph mCT PET/CT scanner (Siemens). A low-dose CT scan was used for attenuation correction of PET data. All appropriate corrections, including time of flow (TOF), were applied and reconstruction performed with OSEM (5 iterations, 21 subsets, 2.0 mm Gaussian filter). Visual analysis of PET scans was performed as well as semi-quantitatively using the syngo.via program. Comparison with healthy individuals was performed with three-dimensional stereotactic surface projection (3D-SSP) software using the whole brain as a reference region as well as an automated volume of interest (VOI)-based analysis of FDG uptake in the cortical and subcortical regions. None of the patients were treated with psychotropic medicines that may alter brain glucose metabolism.
Genetic Analyses
Pathological nucleotide expansions for SCA1, SCA2, SCA3, SCA6, SCA7, SCA8 and dentatorubral-pallidoluysian atrophy (DRPLA) were ruled out first in the index case. In the same patient, a targeted high-throughput next-generation panel based on Illumina HiSeq platform was applied (CeGaT GmbH, Tübingen, Germany). This panel contained 300 genes of which the following are associated with ataxia: ABCB7, ABHD12, ADCK3, AFG3L2, AMACR, ANO10, APTX, ATM, ATN1, ATP8A2, ATP1A3, ATXN1, ATXN2, ATXN3, ATXN7, ATXN10, BEAN1, C10orf2, CACNA1A, CCDC88C, COX20, CYP27A1, DARS2, DNMT1, EEF2, ELOVL4, ELOVL5, FGF14, FXN, GBA2, GJB1, GOSR2, GRID2, ITPR1, KCNC3, KCND3, KIF1C, MRE11A, MTPAP, MTTP, NOP56, NPC1, NPC2, OPA1, OPA3, PDYN, PEX7, PHYH, PNPLA6, POLG, PPP2R2B, PRKCG, SACS, SETX, SIL1, SPG7, SPTBN2, STUB1, SYNE1, TBP, TDP1, TGM6, TTBK2, TTPA, VAMP1, VLDLR, WWOX, and ZNF592. Segregation was performed after a variant in KCND3 was identified in the index case.
Results
Clinical Findings
The clinical features are summarized in Table 1. Age of onset was not possible to determine in the index case (IV:1) who is now 45 years old. She reported clumsiness since early childhood with clear difficulties to perform balance exercises in school. Since age 23 years, the patient has reported a clear progression but was not referred to us until age 40 years. During this period, her speech had become slurred and the patient experienced numbness in her extremities; neurophysiology was normal in two occasions, nevertheless. Patient III:2 is 65 years old; she never sought medical care for her ataxia. She presented with a recurrent no-no head tremor for many years and clumsiness since childhood. Similar to her daughter, she has been unable to perform tandem gait since early age. The exact age of onset was not possible to determine; however, the patient reports unsteadiness for as long as she can remember. Her past medical history (PMH) consisted of type 2 diabetes (T2DM) and hypertension. The rate of ataxia progression was very slow; however, during the last 12 years, she reported several falls and suffered from limb fractures in four different occasions. Last year, she was diagnosed with osteoporosis and started treatment with a bisphosphonate. Increased threshold for cold was found suggesting incipient small fiber neuropathy but the remaining neurophysiology was otherwise normal. Patient III:1 reported a similar age of onset with very slow progression rate. However, ataxia in this 78-year-old man has progressed quickly during the last 2 years. His comorbidities include chronic conditions like T2DM, obesity (BMI = 31), and asthma. Right kidney cancer was diagnosed at age 76 years and motivated nephrectomy; since then, the patient has been on dialysis. Last year, he was admitted to hospital for acute abdominal pain. Intestinal ischemia due to an episode of paroxysmal atrial flutter was diagnosed which necessitated surgery. A severe dysphagia became evident after this procedure, and the patient needed a percutaneous endoscopic gastrostomy (PEG) for 5 months. He is now wheel chair-dependent but is still able to navigate it. Neurological findings at examination included severe ataxia, areflexia, nystagmus, restricted vertical gaze, and increased muscle tone in the left arm. Neurophysiological tests revealed a length-dependent sensory axonal polyneuropathy. The youngest patient (V:1) is 21 years old; he was healthy and able to play American football until age 18 years. At this point, he started to experience truncal tremor and gait difficulties. His tremor was alleviated with ondansetron. His ataxia is mainly axial and has not progressed in the course of 2 years. Besides dysmetria, nystagmus and hypometric saccades were evident upon examination. Reflexes and neurophysiological tests were normal (Table 1).
Patients III:1 and III:2 had limited school attendance. Patient III:1 went to elementary school for 7 years while patient III:2 went to a vocational school (2 years) after 8 years of elementary school. Patients III:1 and III:2 declined psychometric testing for the purpose of this study. Patient III:1 had a MoCA score of 15points with major deficits found in visuospatial/executive tasks as well as in attention and delayed recall. Patient III:2 obtained a MoCA score of 24; this examination revealed mainly deficits in attention. Patient IV:1 has a total education of 13 years and has been working as a nursing assistant, her score was 25. Patient V:1 has totally 12 years of education, and in the last 2 years in school, he went through a vocationally oriented program preparing for work with children and youth. A brief cognitive examination with MMT yielded an average result (28 points) while his MoCA score was 22 which is significantly beneath the normal range. The cognitive deficits in patient IV:1 were mild and non-significant. On the other hand, patient V:1 had a significantly low performance in tests assessing visuospatial episodic memory (ROCFT), working memory (Digit span/WAIS), picture naming ability (BNT), word fluency (FAS/COWAT), and executive function/simultaneous capacity (TMT B). In contrast, the result for verbal episodic long term memory was significantly above average (RAVLT/retention). The cognitive features are summarized in Table 2.
None of the four patients had signs of Brugada syndrome or ventricular arrhythmia; there were no cases of SUDS in this family either. Recently, patient III:1 was found to have a bifascicular block, a long-term registration with an ambulatory electrocardiography device has been ordered and the patient has been referred for a new cardiac risk stratification. A previous long-term registration yielded normal results.
Structural Brain Imaging and [18F] FDG PET
The imaging data are summarized in Table 1 and are shown in Figs. 2, 3, 4, 5, 6, 7, and 8. Brain MRI revealed atrophy of the vermis and supratentorial white matter hyperintensities (WMH) in patients III:1 and III:2 and to a lesser degree in the index case (Figs. 2, 3, and 5). These WMH were particularly widespread in III:1 (Fig. 2). The index case was not affected by any other comorbidity usually associated with WMH, suggesting that these abnormalities may be an underlying feature of SCA19/22. WMH were not evident in the youngest patient (V:1). [18F] FDG PET revealed hypometabolism in the entire cerebellum, thalamus, prefrontal cortex (PFC), and parietal regions of the index case (IV:1) (Fig. 6). Her mother had reduced glucose metabolism not only in the PFC but also in the vermis as wells as in the motor and temporal cortex (Fig. 4). The youngest patient (V:1) had hypometabolism in the PFC, parietal, and inferior temporal regions but surprisingly not in the cerebellum (Fig. 8). In the latter structure, atrophy of the vermis was evident (Fig. 7).

Brain MRI of patient III:1. a Midsagittal T2-weighted image displays moderate vermis atrophy. b Coronal T2-weighted image showing periventricular and deep white matter hyperintensities

Brain MRI of patient III:2. a Mild vermis atrophy is evident on this midsagittal T2-weighted image. b Coronal T2-weighted image displays deep white matter hyperintensities

Surface projections (3DSSP) of brain [18F] FDG PET findings for patient III:2. a Widespread hypometabolism in the prefrontal cortex and some areas of the motor and temporal cortices is evident. Metabolism is also reduced in the vermis but normal in the cerebellar hemispheres. b Midsagittal image shows reduced FDG uptake in the vermis (upper case) as compared to the reference values in VOI templates (lower case). c Coronal image shows normal FDG uptake in the putamen (upper case) as compared to the reference values (lower case)

Brain MRI of patient III:2. a Midsagittal T2-weighted image shows mild vermis atrophy. b Punctate white matter hyperintensities in the frontal lobe are evident in the coronal T2-weighted image

Surface projections (3DSSP) of brain [18F] FDG PET findings for patient IV:1. a Metabolism is reduced in the prefrontal and parietal cortex as well as in the entire cerebellum. b Midsagittal image shows reduced FDG uptake in the cerebellum (upper case) as compared to the reference values in the VOI templates (lower case). c A similar abnormality is evident in the thalami on the coronal image (upper case) as compared to the reference values (lower case), FDG uptake in the putamen is otherwise normal

Brain MRI of patient V:1. a Midsagittal and coronal T1-weighted image. b Mild cerebellar atrophy

Surface projections (3DSSP) of brain [18F] FDG PET findings for patient V:1. a Displays hypometabolism in the prefrontal cortex, lateral temporal cortex, and in some areas of the parietal cortex. b Metabolism in the cerebellum is normal on this midsagittal image (upper case). c FDG uptake is also normal in the basal ganglia and thalami on the coronal image (upper case); reference values in VOI templates are displayed in the lower cases of B and C
Genetic Analyses
The heterozygous T377M (c.1130C>T) mutation in KCND3 was identified in the index case first and confirmed by Sanger sequencing. The other affected in the family (III:1, III:2, and IV:1) were found to harbor this mutation. T377M is located in a highly conserved amino acid in exon 3 that is predicted to be pathogenic by three different programs (Mutation Taster, PolyPhen-2 and SIFT). In addition, this variant is absent in ExAC and Swedgene databases. The latter contains genome data on 1000 subjects. Taken together, the T377M is now a class 5 variant according to current criteria proposed by the American College of Medical Genetics [18].
Discussion
Our findings cement the T377 variant as a causative mutation for SCA19/SCA22. This is the first time that functional imaging data is reported in SCA19/22; also new is the ethnic background of this family. Ataxia channelopathies are in general characterized by early motor onset, intellectual disability and slow disease progression as demonstrated in a recent genetic screening performed in a large European ataxia cohort [12]. Most of our findings are in line with those conclusions. In the aforementioned screening which included 412 patients, only one clear pathogenic variant and two variants of unclear significance (VUS) in the KCND3 gene were identified. [12]. In a large cohort of 1500 ataxia patients in the UK, only three SCA19/SCA22 patients were identified while a previous screening in a smaller Asian cohort yielded negative results [13, 19]. Penetrance of KCND3 mutations associated with ataxia is very high as shown in the Swedish family; reduced penetrance has been described only once for KCND3 mutations (M373I) [6].
The spectrum of KCND3 mutations constitutes a unique cardiocerebral syndrome; KCND3 is the only known ataxia gene associated with cardiac arrhythmia so far. Besides the described associations with SUDS and Brugada syndrome, a recent association with atrial fibrillation has been reported [20]. Accordingly, the KCND3 gene is widely expressed; higher levels of expression are found in the cerebellum and in the heart [21,22,23]. Despite the importance of voltage-gated K+ (Kv) channels for action potential repolarization in cardiomyocytes, KCND3 knockout mice do not differ from wild type animals at least in terms of survival as well as for structural and electrophysiological properties of cardiomyocytes [24]. To our knowledge, there is no published data on any potential structural or functional cerebellar abnormalities in this knockout model. Of all the four mutations or variants in KCND3 associated with Brugada syndrome or SUDS, three are located in the C-terminal and one in the S6 transmembrane domain A. [9,10,11]. This scarcity of cases precludes the delineation of clear genotype-phenotype correlations at the moment; however, risk stratification for cardiac arrhythmia is warranted in patients with KCND3 mutations regardless of the mutation type or location within the gene. Other familial potassium channelopathies associated with cerebellar dysfunction include spinocerebellar ataxia type 13 (SCA13), episodic ataxia type 1 (EA1), and sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SESAME syndrome) which are caused by mutations in the KCNC3, KCNA1, and KCNJ10 genes, respectively [25,26,27]. Different to SCA13 in which some patients have a non-progressive course [28], SCA19/22 is clearly progressive [6, 7]. Interestingly, an association between epilepsy and long QT syndrome (LQTS) has been proposed for mutations in KCNQ1; however, ataxia is, to the best of our knowledge, not part of the disease spectrum associated with KCNQ1 mutations [29]. Ataxia is on the other hand one of several acquired autoimmune disorders in the spectrum associated with antibodies to voltage-gated potassium channel (VGKC) which also include Morvan’s syndrome, neuromyotonia, limbic encephalitis, and cardiac arrhythmia [30,31,32].
All the four patients in the Swedish SCA19/22 family display atrophy of the vermis and three of them display varying degrees of white matter hyperintensities (WMH). WMH are common findings among the elderly and of vascular origin; these abnormalities are associated with cognitive decline particularly when they are progressive [33, 34]. Different to the previously described SCA19/22 families, we did not find clear atrophy in the cerebellar hemispheres or the less common cerebral atrophy [4, 35]. In contrast to a Dutch SCA19 family, we did not find evidence of myoclonus nor psychiatric manifestations (e.g., impulsivity or depression) [36]. Onset in the index case and patient III:2 in the Swedish family was during early childhood, but the exact timing was not possible to determine. Early onset is in fact very rare for SCA19/22, but slow disease progression is common [15, 36]. At least in one of the patients (III:1), there was evidence of global cognitive impairment as evaluated by MoCA. These deficits were predominantly in visuospatial and executive tasks (IV:1 and V:1) and are similar to what have been reported in two unrelated French families [14]. However, the French families display a more complex syndrome that includes Parkinsonism, seizures, as well as psychiatric symptoms. Previously, impairment in the Wisconsin Cart Sorting Test (WCST) suggesting impaired ability for abstract thinking and set-shifting was identified in SCA19 [14, 37, 36]. Similar deficits have been found in some of the polyglutamine SCAs (SCA1, SCA2, SCA3, and SCA6) [38, 39]. The cognitive deficits we have found in this family and their pattern of brain glucose metabolism support the notion that a frontal-executive dysfunction occurs in SCA19 [36]. It is noticeable that the general intellectual ability was not low neither in patient IV:1 nor V:1. However, the contrast between patient IV:1 whose cognitive deficits are rather mild and the pronounced dysfunction in several cognitive domains found in patient V:1 is striking. Mild deficits are hard to reconcile with the widespread brain hypomethabolism in patient IV:1. The reasons for this remarkable intrafamilial variability are unknown at the moment. The main limitations of this study are small sample size and the fact that neuropsychological testing and [18F] FDG PET were performed at different time points.
Reasonably, significant comorbidity contributed to the faster rate of motor progression and low MoCA score in the oldest patient (III:1) in the Swedish SCA19/22 family. Both T2DM and kidney failure are well-known risk factors for cognitive decline and dementia [40, 41]. Differently to the described Japanese SCA19/22 patient harboring the T377M mutation in KCND3, we found polyneuropathy in two patients from the Swedish family who were also affected by T2DM. However, this feature has to be interpreted with great caution since damage to periphery nerves is commonly associated with T2DM. Nonetheless, it will be of value to learn more about non-ataxia features in relatives to the Japanese patient harboring the T377M mutation [7].
Few PET studies have been performed in SCA; so far, only two small [18F] FDG PET studies on ataxia channelopathies have been published [42, 43]. The pattern of hypometabolism we described in SCA19/22 is reminiscent of the abnormal pattern found mainly in SCA6 and to some degree in SCA2 [43]. In SCA6, a calcium channelopathy, reduced glucose metabolism was found in the cerebellum and frontal and prefrontal cortices [43]. Different to SCA3 we did not find any evidence of abnormal metabolism in the basal ganglia. Our findings regarding variable brain hypometabolism are not specific enough to draw definitive conclusions. Additional studies are needed in order to assess the pattern of FDG metabolism in patients with other KCND3 mutations.
Only one SCA19/22 neuropathological assessment has been reported so far; besides moderate to severe loss of Purkinje cells, reduced levels of KCND3 protein were found in cerebellar homogenates [44]. Also similar to SCA6, some neuronal loss was identified in a few brainstem nuclei [3, 45]. Some of the mutant Kv4.3 proteins remain trapped in the endoplasmic reticulum leading to a loss of function [6, 7]. Later, evidence of a dominant negative effect in SCA19/22 was provided for some mutations [10, 11] which is similar to previous findings in EA1 and SCA13 [26, 27, 46, 47]. Despite these important advances, many aspect of the disease remain to be explored. For instance, long-term follow-up studies are needed to determine the prevalence and risk of cardiac arrhythmia. Additional neuropathological studies of SCA19/22 are needed in order to establish genotype-phenotype and clinic-pathological correlations. Finally, the potential pathogenic role of the two recently identified VUS in KCND3 [12] and the impact of potential gene modulators on disease expressivity remain also to be investigated.
References
Depondt C, Donatello S, Rai M, Wang FC, Manto M, Simonis N, et al. MME mutation in dominant spinocerebellar ataxia with neuropathy (SCA43). Neurology Genetics. 2016;2(5):e94. https://doi.org/10.1212/NXG.0000000000000094.
Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. The Lancet. Neurology. 2010;9(9):885–94. https://doi.org/10.1016/S1474-4422(10)70183-6.
Rüb U, Schöls L, Paulson H, Auburger G, Kermer P, Jen JC, et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol. 2013a;104:38–66. https://doi.org/10.1016/j.pneurobio.2013.01.001.
Chung M-Y, Lu Y-C, Cheng N-C, Soong B-W. A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain J Neurol. 2003;126(Pt 6):1293–9.
Verbeek DS, Schelhaas JH, Ippel EF, Beemer FA, Pearson PL, Sinke RJ. Identification of a novel SCA locus ( SCA19) in a Dutch autosomal dominant cerebellar ataxia family on chromosome region 1p21-q21. Hum Genet. 2002;111(4–5):388–93. https://doi.org/10.1007/s00439-002-0782-7.
Duarri A, Jezierska J, Fokkens M, Meijer M, Schelhaas HJ, den Dunnen WFA, et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann Neurol. 2012;72(6):870–80. https://doi.org/10.1002/ana.23700.
Lee YC, Durr A, Majczenko K, Huang Y-H, Liu Y-C, Lien C-C, et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol. 2012;72(6):859–69. https://doi.org/10.1002/ana.23701.
Pulst S-M, Otis TS. Repolarization matters: mutations in the Kv4.3 potassium channel cause SCA19/22. Ann Neurol. 2012;72(6):829–31. https://doi.org/10.1002/ana.23803.
Giudicessi JR, Ye D, Kritzberger CJ, Nesterenko VV, Tester DJ, Antzelevitch C, et al. Novel mutations in the KCND3-encoded Kv4.3 K+ channel associated with autopsy-negative sudden unexplained death. Hum Mutat. 2012;33(6):989–97. https://doi.org/10.1002/humu.22058.
Duarri A, Lin M-CA, Fokkens MR, Meijer M, Smeets CJLM, Nibbeling EAR, et al. Spinocerebellar ataxia type 19/22 mutations alter heterocomplex Kv4.3 channel function and gating in a dominant manner. Cell Mol Life Sci. 2015a;72(17):3387–99. https://doi.org/10.1007/s00018-015-1894-2.
Duarri A, Nibbeling E, Fokkens MR, Meijer M, Boddeke E, Lagrange E, et al. Erratum to: the L450F [corrected] mutation in KCND3 brings spinocerebellar ataxia and Brugada syndrome closer together. Neurogenetics. 2015b;16(3):243. https://doi.org/10.1007/s10048-015-0447-z.
Coutelier M, Coarelli G, Monin M-L, Konop J, Davoine C-S, Tesson C, et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain. 2017; https://doi.org/10.1093/brain/awx081.
Hadjivassiliou M, Martindale J, Shanmugarajah P, Grünewald RA, Sarrigiannis PG, Beauchamp N, et al. Causes of progressive cerebellar ataxia: prospective evaluation of 1500 patients. J Neurol Neurosurg Psychiatry. 2017;88(4):301–9.
Huin V, Strubi-Vuillaume I, Dujardin K, Brion M, Delliaux M, Dellacherie D, et al. Expanding the phenotype of SCA19/22: parkinsonism, cognitive impairment and epilepsy. Parkinsonism Relat Disord. 2017;45:85–9.
Smets K, Duarri A, Deconinck T, Ceulemans B, van de Warrenburg BP, Züchner S, et al. First de novo KCND3 mutation causes severe Kv4.3 channel dysfunction leading to early onset cerebellar ataxia, intellectual disability, oral apraxia and epilepsy. BMC Med Genet. 2015;16:51. https://doi.org/10.1186/s12881-015-0200-3.
Lezak, M. D. (n.d.2012). Neuropsychological assessment, (5th ed.). New York: Oxford University Press.
Strauss, E. (n.d.). Compendium of neuropsychological tests: administration, norms, and commentary. Appl Neuropsychol 2007;14(1):62–63.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Choubtum L, Witoonpanich P, Hanchaiphiboolkul S, Bhidayasiri R, Jitkritsadakul O, Pongpakdee S, et al. Analysis of SCA8, SCA10, SCA12, SCA17 and SCA19 in patients with unknown spinocerebellar ataxia: a Thai multicentre study. BMC Neurol. 2015;15:166. https://doi.org/10.1186/s12883-015-0425-y.
Low S-K, Takahashi A, Ebana Y, Ozaki K, Christophersen IE, Ellinor PT, et al. Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat Genet. 2017;49:953–8. https://doi.org/10.1038/ng.3842.
Hsu Y-H, Huang H-Y, Tsaur M-L. Contrasting expression of Kv4.3, an A-type K+ channel, in migrating Purkinje cells and other post-migratory cerebellar neurons. Eur J Neurosci. 2003;18(3):601–12.
Isbrandt D, Leicher T, Waldschütz R, Zhu X, Luhmann U, Michel U, et al. Gene structures and expression profiles of three human KCND (Kv4) potassium channels mediating A-type currents I(TO) and I(SA). Genomics. 2000;64(2):144–54. https://doi.org/10.1006/geno.2000.6117.
Tsaur ML, Chou CC, Shih YH, Wang HL. Cloning, expression and CNS distribution of Kv4.3, an A-type K+ channel alpha subunit. FEBS Lett. 1997;400(2):215–20.
Niwa N, Wang W, Sha Q, Marionneau C, Nerbonne JM. Kv4.3 is not required for the generation of functional Ito, f channels in adult mouse ventricles. J Mol Cell Cardiol. 2008;44(1):95–104. https://doi.org/10.1016/j.yjmcc.2007.10.007.
Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360(19):1960–70. https://doi.org/10.1056/NEJMoa0810276.
D’Adamo MC.. Episodic Ataxia Type 1. In R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, L. et al (Eds.), GeneReviews(®). Seattle (WA): University of Washington, Seattle. 1993. Retrieved from http://www.ncbi.nlm.nih.gov/books/NBK25442.
Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JPA, Nolte D, et al. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38(4):447–51. https://doi.org/10.1038/ng1758.
Khare S, Nick JA, Zhang Y, Galeano K, Butler B, Khoshbouei H, et al. A KCNC3 mutation causes a neurodevelopmental, non-progressive SCA13 subtype associated with dominant negative effects and aberrant EGFR trafficking. PLoS One. 2017;12(5):e0173565. https://doi.org/10.1371/journal.pone.0173565.
Tiron C, de Llano CT, Campuzano O, Pérez-Serra A, Mademont I, Coll M, et al. Further evidence of the association between LQT syndrome and epilepsy in a family with KCNQ1 pathogenic variant. Seizure. 2015;25:65–7. https://doi.org/10.1016/j.seizure.2015.01.003.
Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008;7(4):327–40. https://doi.org/10.1016/S1474-4422(08)70060-7.
Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain J Neurol. 2010;133(9):2734–48. https://doi.org/10.1093/brain/awq213.
Liguori R, Vincent A, Clover L, Avoni P, Plazzi G, Cortelli P, et al. Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain J Neurol. 2001;124(Pt 12):2417–26.
Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ (Clinical Research Ed). 2010;341:c3666.
Kloppenborg RP, Nederkoorn PJ, Geerlings MI, van den Berg E. Presence and progression of white matter hyperintensities and cognition: a meta-analysis. Neurology. 2014;82(23):2127–38. https://doi.org/10.1212/WNL.0000000000000505.
Schelhaas HJ, Ippel PF, Hageman G, Sinke RJ, van der Laan EN, Beemer FA. Clinical and genetic analysis of a four-generation family with a distinct autosomal dominant cerebellar ataxia. J Neurol. 2001;248(2):113–20.
Schelhaas HJ, van de Warrenburg BPC. Clinical, psychological, and genetic characteristics of spinocerebellar ataxia type 19 (SCA19). Cerebellum (London, England). 2005;4(1):51–4. https://doi.org/10.1080/14734220510007888.
Schelhaas HJ, van de Warrenburg BPC, Hageman G, Ippel EE, van Hout M, Kremer B. Cognitive impairment in SCA-19. Acta Neurol Belg. 2003;103(4):199–205.
Bürk K, Globas C, Bösch S, Klockgether T, Zühlke C, Daum I, et al. Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J Neurol. 2003;250(2):207–11. https://doi.org/10.1007/s00415-003-0976-5.
Globas C, Bösch S, Zühlke C, Daum I, Dichgans J, Bürk K. The cerebellum and cognition. Intellectual function in spinocerebellar ataxia type 6 (SCA6). J Neurol. 2003;250(12):1482–7. https://doi.org/10.1007/s00415-003-0258-2.
Kurella Tamura M, Tam K, Vittinghoff E, Raj D, Sozio SM, Rosas SE, et al. Inflammatory markers and risk for cognitive decline in chronic kidney disease: the CRIC study. Kidney Int Rep. 2017;2(2):192–200. https://doi.org/10.1016/j.ekir.2016.10.007.
Moran, C., Beare, R., Phan, T. G., Bruce, D. G., Callisaya, M. L., Srikanth, V., (ADNI). et al (2015). Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology, 85(13), 1123–1130. doi:https://doi.org/10.1212/WNL.0000000000001982.
Soong B, Liu R, Wu L, Lu Y, Lee H. Metabolic characterization of spinocerebellar ataxia type 6. Arch Neurol. 2001;58(2):300–4.
Wang P-S, Liu R-S, Yang B-H, Soong B-W. Regional patterns of cerebral glucose metabolism in spinocerebellar ataxia type 2, 3 and 6: a voxel-based FDG-positron emission tomography analysis. J Neurol. 2007;254(7):838–45. https://doi.org/10.1007/s00415-006-0383-9.
Seidel K, Küsters B, den Dunnen WFA, Bouzrou M, Hageman G, Korf H-W, et al. First patho-anatomical investigation of the brain of a SCA19 patient. Neuropathol Appl Neurobiol. 2014;40(5):640–4. https://doi.org/10.1111/nan.12128.
Seidel K, Siswanto S, Brunt ERP, den Dunnen W, Korf H-W, Rüb U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012;124(1):1–21. https://doi.org/10.1007/s00401-012-1000-x.
Minassian NA, Lin M-CA, Papazian DM. Altered Kv3.3 channel gating in early-onset spinocerebellar ataxia type 13. J Physiol. 2012;590(7):1599–614. https://doi.org/10.1113/jphysiol.2012.228205.
Zerr P, Adelman JP, Maylie J. Episodic ataxia mutations in Kv1.1 alter potassium channel function by dominant negative effects or haploinsufficiency. J Neurosci. 1998;18(8):2842–8.
Acknowledgements
Our gratitude is to the patients for their kind participation in this study. Also, thanks to Mariann Skoglund for performing one of the psychometric evaluations and to Professor Chantal Tallaksen for providing valuable opinions on the draft.
Funding
Martin Paucar was supported by the Stockholm County Council.
Author information
Authors and Affiliations
Contributions
M. Paucar designed the study and wrote the first draft. I. Savitcheva analyzed the neuroimaging data. Å. Bergendahl wrote part of the first draft by summarizing the cognitive data. J. Laffita reviewed the data and the first draft version. P. Gustavsson and M. Nordenskjöld review of the genetic data and draft. P. Svenningsson contributed to the discussion on study design, interpretation of data, and critical review of the first draft.
Corresponding author
Ethics declarations
This study was carried out in accordance with the recommendations of the ethics committee in Stockholm and the radiation protection organization at the Karolinska University Hospital (Etikprövningsnämnden dnr 2016/2538-32) with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the ethics committee in Stockholm.
Conflict of Interest
The authors declare that there is no conflict of interest.
Electronic Supplementary Material
Supplementary material
Variable degree of ataxia, nystagmus and dysarthria are evident in three patients with the T377M mutation in KCND3, these recordings made in 2014 and 2015. The oldest patient in this kindred had the most severe motor features; axial ataxia predominates but variable dysmetria is also evident in all the three patients (the youngest patient is not shown). (MP4 272494 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Paucar, M., Bergendal, Å., Gustavsson, P. et al. Novel Features and Abnormal Pattern of Cerebral Glucose Metabolism in Spinocerebellar Ataxia 19. Cerebellum 17, 465–476 (2018). https://doi.org/10.1007/s12311-018-0927-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-018-0927-4