
Abstract
Per the 2017 WHO, several translocations have been described that are sufficient for the diagnosis of acute myeloid leukemia with myelodysplasia-related changes (AML-MRC) (assuming no prior therapy and ≥ 20% myeloblasts present in blood or bone marrow), including the t(1;3)(p36;q21). This translocation juxtaposes the RPN1 gene (3q21.2) promoter upstream of the PRDM16 gene (1p36) resulting in PRDM16 overexpression. While uncommon, PRDM16 overexpression is considered an unfavorable prognostic finding in myeloid neoplasms. A variant PRDM16 rearrangement t(1;2)(p36;p21) has been rarely described in various hematologic neoplasms, including two cases of myelodysplastic syndrome and one case each of myelofibrosis and T-lymphoblastic leukemia. We describe the first case to our knowledge of t(1;2)(p36;p21) observed in AML-MRC. In addition, a next-generation sequencing strategy, mate-pair sequencing (MPseq) was performed and revealed the promoter 2 region of THADA (2p21) was juxtaposed upstream from PRDM16 which may be responsible for PRDM16 overexpression that has been reported in hematologic neoplasms harboring the t(1;2)(p36;p21).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recurrent balanced chromosomal rearrangements in acute myeloid leukemia (AML) have diagnostic-, prognostic-, and/or treatment-related significance and the majority result in gene fusions that encode chimeric proteins [1]. While less commonly observed in AML with myelodysplasia-related changes (AML-MRC), several reciprocal translocations have been described that are sufficient for the diagnosis of AML-MRC (assuming no prior therapy and ≥ 20% myeloblasts present in blood or bone marrow), including the t(1;3)(p36;q21) [2]. Instead of creating a gene fusion, the 1;3 translocation juxtaposes the RPN1 gene (3q21) promoter upstream of the PRDM16 gene (1p36), resulting in aberrant transcriptional upregulation of PRDM16 [3, 4]. Highly homologous to the MECOM gene region (3q26), PRDM16 has been shown to play a critical role in hematopoietic stem cell maintenance and renewal and is overexpressed in leukemic cells [3,4,5]. Furthermore, overexpression of the PRDM16 gene has been associated with an unfavorable prognosis in myeloid neoplasms [4, 6, 7].
A rarely described PRDM16 translocation variant t(1;2)(p36;p21) is hypothesized to result in PRDM16 overexpression due to the juxtaposition of regulatory elements from 2p21, specifically THADA [4, 8]. To date, only four cases have been described that harbor t(1;2)(p36;p21), including two cases of myelodysplastic syndrome (MDS) and one case each of myelofibrosis and T-lymphoblastic leukemia (T-ALL) [4, 8, 9]. We describe the first case to our knowledge of a t(1;2)(p36;p21) observed in AML-MRC. To further characterize this rearrangement, we utilized a next-generation sequencing strategy, mate-pair sequencing (MPseq), to define the translocation breakpoints with greater resolution and precision compared to traditional cytogenetic methodologies.
Clinical history
A 35-year-old female underwent a bone marrow biopsy for pancytopenia evaluation. Hematologic data obtained from a peripheral blood specimen included a hemoglobin of 5 g/dL (12.0–15.5 g/dL), hematocrit of 14.7% (34.9–44.5%), red blood cell count of 1.2 × 1012/L (3.90–5.03 × 1012/L), white blood cell count of 1.86 × 109/L (3.5–10.5 × 109/L), and platelet count of 72 × 109/L (150–450 × 109/L). The peripheral blood smear revealed macrocytic normochromic red cells with marked absolute neutropenia, a rare circulating blast, and moderately decreased platelets. The bone marrow aspirate smears showed decreased erythroid precursors with nuclear irregularity and asynchronous maturation, and decreased granulopoiesis with hypogranular and abnormal segmentation of the neutrophils. The bone marrow biopsy was mildly hypocellular (30–40%) with 16% myeloid blasts, 6% promonocytes, 17% monocytes, and increased numbers of dysplastic appearing megakaryocytes, several of which were small and hypolobated. Immunohistochemistry with CD34 demonstrated overall positivity in approximately 20% of nuclei with occasional small clusters. Flow cytometry of the bone marrow revealed 10% myeloid blasts [positive for CD117, CD34, HLA-DR (variable), CD33 (dim partial), CD13 (dim partial), and CD64 (dim partial)] and 17% monocytes with an aberrant immunophenotype [positive for CD14 (variable), CD64, HLA-DR, CD33, and CD13 (partial and variable)]. Based on these findings, a diagnosis of AML-MRC was rendered.
Materials and methods
Conventional chromosome and fluorescence in situ hybridization studies
All genomic studies were performed on the diagnostic bone marrow aspirate specimen. For conventional chromosome studies, G-banding by trypsin using Leishman stain was performed on bone marrow cells that were cultured and harvested as per protocol. A total of 20 metaphases were fully analyzed. The bone marrow aspirate specimen was processed for fluorescence in situ hybridization (FISH) according to specimen-specific laboratory protocols and subjected to standard pretreatment, hybridization, and fluorescence microscopy. A “laboratory developed” PRDM16/RPN1 dual-color dual-fusion (D-FISH) probe set was utilized. Conventional chromosome and FISH results were interpreted by a board-certified clinical cytogeneticist (ABMGG).
Mate-pair sequencing
DNA was processed using Illumina Nextera Mate Pair library kit (Illumina, San Diego, CA), multiplexed at two samples per lane, and sequenced on the Illumina HiSeq 2500 using 101-basepair reads and paired end sequencing in Rapid Run mode. Data were aligned to the reference genome (GRCh38) using BIMAv3, and abnormalities were identified and visualized using SVAtools and Ingenium, both in-house developed bioinformatics tools [10, 11].
Results
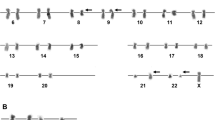
Conventional chromosome studies identified an apparently balanced t(1;2)(p36.1;p21) in all 20 metaphases analyzed (Fig. 1a). The PRDM16/RPN1 D-FISH probe set did not demonstrate fusion of these two probes, but revealed a third (additional) PRDM16 signal in 413 (82.6%) of 500 interphase nuclei (Fig. 1b). Sequential FISH studies with the PRDM16/RPN1 D-FISH probe set indicated the three PRDM16 signals were located on the normal chromosome 1p, on the derivative chromosome 1p, and on the derivative chromosome 2p (Fig. 1c).

Representative conventional chromosome and FISH images. a Karyogram demonstrating a t(1;2)(p36.1;p21). This translocation was observed in all 20 metaphases analyzed. b Representative interphase nuclei demonstrating three PRDM16 signals (red) indicated by arrows. This signal pattern was observed in 413 (82.6%) of 500 interphase nuclei. c Sequential FISH studies using the PRDM16/RPN1 D-FISH probe set illustrating the locations of the three PRDM16 signals (red). The PRDM16 FISH probe hybridized to the normal chromosome 1p and at the translocation breakpoints on the derivative chromosomes 1p and 2p (arrows)
To further interrogate the 1;2 translocation, MPseq was performed and characterized the translocation between 1p36.32 and 2p21. This translocation did not result in a chimeric fusion gene (Fig. 2); however, this rearrangement juxtaposed the promoter 2 region of the THADA gene approximately 267 kb upstream from the PRDM16 gene region (Fig. 3). Sanger sequencing subsequently confirmed the translocation breakpoints identified by MPseq (Table 1). While this translocation did not disrupt the PRDM16 gene, the 1p36.32 breakpoint “disrupted” the PRDM16 FISH probe footprint resulting in a PRDM16 FISH signal located at the translocation breakpoints on each of the derivative chromosomes 1p and 2p (Fig. 3).

Mate-pair sequencing (MPseq) results visualized in SVAtools. Junction plot demonstrating a translocation between 1p36.32 and 2p21. Note that while both breakpoints involved intergenic regions, the PRDM16 FISH probe was disrupted (horizontal red bar)

A focused view of the t(1;2)(p36.32;p21). Dashed horizontal black lines indicate breakpoints on the derivative chromosomes 1 and 2. This translocation juxtaposes the promoter 2 region of THADA upstream from PRDM16. The translocation breakpoint on 1p is also consistent with a disruption of the PRDM16 FISH probe and a translocation of a portion of the PRDM16 FISH probe to the derivative 2p, as observed by PRDM16/RPN1 D-FISH studies
Discussion
Upregulation of the PRDM16 gene by promoter swap is a distinct entity in myeloid neoplasms and is associated with an unfavorable prognosis [4]. While the t(1;3)(p36;q21) is the most common rearrangement associated with PRDM16 upregulation, four cases of hematologic neoplasms have been described in the literature that harbor the rare variant, t(1;2)(p36;p21). Duhoux et al described a 63-year-old male with myelofibrosis and a 79-year-old female with T-ALL, each harboring a t(1;2) that was postulated to involve the THADA gene region (as defined by FISH studies) [4]. Moreover, overexpression of the PRDM16 gene was confirmed in the case of T-ALL by gene expression studies. Similarly, a t(1;2) involving the THADA gene region by FISH was also observed in a 70-year-old female with MDS that resulted in upregulation of PRDM16 by gene expression studies [8]. Lastly, Masuya et al described a similar t(1;2) in a 55-year-old female with therapy-related MDS, although neither FISH nor PRDM16 gene expression studies were performed [9]. In addition, while only evaluated thus far in a single patient, gene expression of THADA may not be compromised due to the t(1;2) as demonstrated by gene expression studies performed on the 70-year-old female with MDS [8].
Adding to the literature, we are describing the youngest patient harboring a t(1;2)(p36;p21), a 35-year-old female with a diagnosis of AML-MRC. We verified a disruption of the PRDM16 gene region by FISH analysis (Fig. 1), performed MPseq, and revealed a translocation between intergenic chromosomal regions 1p36.32 and 2p21 (Fig. 2). While a fusion gene was not created, promoter 2 of THADA was juxtaposed 267 kb upstream from PRDM16 (Fig. 3). Although PRDM16 gene expression studies were not performed on our case, MPseq results indicate the promoter 2 region of THADA likely represents the previously uncharacterized regulatory element within the 2p21 chromosome region that is responsible for PRDM16 overexpression in these rare cases with a t(1;2)(p36;p21). While MPseq further characterized this translocation, conventional chromosome analysis would be sufficient to detect and monitor a hematologic neoplasm harboring the t(1;2)(p36;p21).
In summary, we further characterized t(1;2)(p36;p21) involving the PRDM16 and THADA gene regions by MPseq in a newly diagnosed case of AML-MRC. Although PRDM16 gene expression studies were not performed, juxtaposition of the promoter 2 region of THADA upstream of the PRDM16 gene is likely responsible for PRDM16 overexpression that has been reported in the various hematologic neoplasms harboring a t(1;2)(p36;p21) [4, 8].
References
Arber DA, Brunning RD, Le Beau MM et al (2017) Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL et al (eds) WHO classification of tumours of haematopoietic and lymphoid tissues, revised 4th edn. IARC, Lyon, pp 130–149
Arber DA, Brunning RD, Orazi A et al (2017) Acute myeloid leukaemia with myelodysplasia-related changes. In: Swerdlow SH, Campo E, Harris NL et al (eds) WHO classification of tumours of haematopoietic and lymphoid tissues, revised 4th edn. IARC, Lyon, pp 150–152
Mochizuki N, Shimizu S, Nagasawa T, Tanaka H, Taniwaki M, Yokota J, Morishita K (2000) A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood 96:3209–3214
Duhoux FP, Ameye G, Montano-Almendras CP et al (2011) PRDM16 (1p36) translocations define a distinct entity of myeloid malignancies with poor prognosis but my also occur in lymphoid malignancies. Br J Haematol 156:76–88
Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, Lemischka IR, Zhou BY, Snoeck HW (2011) Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood 117:5057–5066
Eveillard M, Delaunay J, Richebourg S, Lode L, Garand R, Wuilleme S, Duhoux F, Antoine-Poirel H, Godon C, Bene MC (2015) The closely related rare and severe acute myeloid leukemias carrying EVI1 or PRDM16 rearrangements share singular biological features. Haematologica 100:e114–e115
Yamato G, Yamaguchi H, Handa H, Shiba N, Kawamura M, Wakita S, Inokuchi K, Hara Y, Ohki K, Okubo J, Park MJ, Sotomatsu M, Arakawa H, Hayashi Y (2017) Clinical features and prognostic impact of PRDM16 expression in adult acute myeloid leukemia. Genes Chromosom Cancer 56:800–809
Storlazzi CT, Albano F, Guastadisegni MC, Impera L, Mühlematter D, Meyer-Monard S, Wuillemin W, Rocchi M, Jotterand M (2008) Upregulation of MEL1 and FLJ42875 genes by position effect resulting from a t(1;2)(p36;p21) occurring during evolution of chronic myelomonocytic leukemia. Blood Cells Mol Dis 40:452–455
Masuya M, Katayama N, Inagaki K et al (2001) Two independent clones in myelodysplastic syndrome following treatment of acute myeloid leukemia. Int J Hematol 75:182–186
Drucker TM, Johnson SH, Murphy SJ, Cradic KW, Therneau TM, Vasmatzis G (2014) BIMA V3: an aligner customized for mate pair library sequencing. Bioinformatics 30:1627–1629
Johnson SH, Smadbeck JB, Smoley SA, Gaitatzes A, Murphy SJ, Harris FR, Drucker TM, Zenka RM, Pitel BA, Rowsey RA, Hoppman NL, Aypar U, Sukov WR, Jenkins RB, Feldman AL, Kearney HM, Vasmatzis G (2018) SVAtools for junction detection of genome-wide chromosomal rearrangements by mate-pair sequencing (MPseq). Cancer Genet 221:1–18
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
PM, DLVD, MKB, BAP, SAS, JBS, WRS, PTG, RPK, LBB, and JFP declare that they have no conflict of interest. GV: Algorithms described in this manuscript for mate-pair sequencing are licensed to WholeGenome LLC owned by GV.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Muppa, P., Van Dyke, D.L., Bianco, M.K. et al. Characterization of a t(1;2)(p36;p21) involving the PRDM16 gene region by mate-pair sequencing (MPseq) in a patient with newly diagnosed acute myeloid leukemia with myelodysplasia-related changes. J Hematopathol 12, 85–90 (2019). https://doi.org/10.1007/s12308-019-00348-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-019-00348-w