
Abstract
Infantile disseminated BCG-osis is an uncommon complication of BCG vaccination and the presence of haemophagocytic lymphohistiocytosis (HLH) further complicates the clinical course due to its fatal outcome. Here, we describe a rare case of disseminated BCG-osis with HLH in a 3-month-old male child and the unusual morphological findings in the peripheral blood with its haematology analyser-based expression. The child presented with fever, failure to thrive, hepatosplenomegaly, erythematous skin rashes, and left axillary lymphadenopathy with history of BCG vaccination at birth. He was the first born of second-degree consanguineous marriage with no significant family history of immunodeficiency disorders. Laboratory findings included anaemia, thrombocytopenia, hyperferritinaemia, hypertriglyceridaemia, and hypofibrinogenaemia which supported a diagnosis of HLH. The peripheral blood showed evidence of phagocytosis by neutrophils, pseudo-Chediak-Higashi-like inclusions, blue-green inclusions, and intra-cytoplasmic vacuoles with shadowy appearance and cellular debris in the background. Acid-fast bacilli were demonstrated in the peripheral blood by Ziehl-Neelsen stain. His clinical condition gradually worsened with multi organ failure and fatality.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bacille Calmette-Guérin (BCG), a live attenuated Mycobacterium bovis strain vaccine, is routinely given to newborns for prevention of disseminated tuberculosis with almost 90% coverage worldwide [1]. The BCG vaccination is safe with very few adverse events. Mild localised reactions or adenitis are the most commonly reported complications. The more serious, systemic, or disseminated disease involving one or more anatomical sites are very rare with a reported frequency of less than 5 per 1 million vaccinees. The immunocompromised children are more susceptible for disseminated BCG-osis. Specifically, the HIV infection poses a considerable risk of distant or disseminated disease in infants with a high case fatality rate of more than 70% [2]. In non-HIV-infected children, primary immunodeficiency diseases (PID) are more commonly associated with systemic manifestations [3]. Haemophagocytic lymphohistiocytosis (HLH) is an unusual complication of disseminated tuberculosis or BCG-osis with a very high mortality [4]. We report here a rare case of fatal infantile disseminated BCG-osis with haemophagocytic lymphohistiocytosis and its unusual haematological findings. It is more interesting to note the haemocytometric pattern of expression by the automated analyser and the corresponding cell population data (CPD).
Case report
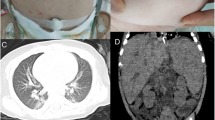
A 3-month-old male child (Fig. 1a, b) presented with fever, abdominal distension, and an ulcerative lesion over left axilla since 3 weeks. He was the first born of second-degree consanguineous marriage with no significant history of any specific primary immunodeficiency diseases in the family. Antenatal history was uneventful. He was delivered by normal vaginal delivery at term with the birth weight appropriate for gestational age. He belonged to middle socioeconomic status with no history of contact with tuberculosis or overcrowding. He was exclusively breast fed without any specific dietary deficiencies. He had received BCG and hepatitis vaccines at birth. The physical examination revealed only pallor without any icterus, cyanosis, or clubbing. A large lymph node measuring 6 cm × 8 cm was present over left axilla with an overlying ulcer. Confluent erythematous patches were seen all over the body with scaling and sparse depigmented hairs. Features of failure to thrive were noted. No facial dysmorphism or any specific anomalies were noted. BCG scar was present on the left arm. Liver was 7 cm enlarged with spleen 9 cm below the left costal margin. Child had poor activity, cry, reflexes, and tone with anterior fontanelle at normal level. The chest x-ray was normal. An ultrasonography showed hepatosplenomegaly and conglomerated matted left axillary lymph nodes measuring 8–10 mm each without any obvious sinus tract.

a, b A 3-month-old male infant with erythematous rashes, splenomegaly (marked on abdomen), and left axillary lymph node with overlying ulcer (pointed by the arrow)
The peripheral blood showed anaemia and thrombocytopenia with leukemoid reaction (haemoglobin 8.5 g/dL, WBC count 43.1 × 103/μL, platelet count 79 × 103/μL). The erythrocyte sedimentation rate (ESR) was elevated with 86 mm in first hour. The C-reactive protein (CRP) was 3.44 mg/dL. The liver function tests showed elevated levels of SGOT (76.6 U/L) and alkaline phosphatase (207.7 U/L). He had hyponatraemia (Na+ 131 mEq/L), hypocalcaemia (Ca2+ 7.5 mg/dL), and hypoproteinaemia (total protein levels 6.4 mg/dL) with hypoalbuminaemia (2.6 mg/dL). The gamma globulin fractions were within normal limits (1.36 g/dL). The screening for HIV, hepatitis B, and hepatitis C was negative. Aerobic cultures for blood, urine, and CSF were negative. A CSF examination elicited mild pleocytosis (total cell count 30/μL) with predominantly mononuclear cells (85%) and normal biochemical findings. Gastric aspirate was positive for acid-fast bacilli (AFB) by Ziehl-Neelsen (Z-N) staining.
The subsequent peripheral blood revealed reducing trend in haemoglobin levels with a nadir of 6.1 g/dL on day 4 while there was an increasing leukocyte count up to the third day followed by a drop in the counts. But surprisingly, the platelet count showed marked thrombocytosis of 929 × 103/μL on the fourth day. When the peripheral smear was reviewed, it showed a leukoerythroblastic blood picture with many striking morphological findings. Those included moderate eosinophilia with eosinophilic vacuoles, neutrophilic leukocytosis with a left shift and toxic changes, granulocytes showing phagocytosis of apoptotic cellular debris (Fig. 2a), large round bluish violet Chediak-Higashi-like inclusions (Fig. 2b), and blue-green cytoplasmic inclusions (Fig. 2c). The background showed a lot of apoptotic and necrotic cellular debris, which were spuriously counted as platelets by the automated cell counter giving rise to a pseudo-thrombocytosis (Fig. 2d). In addition to these findings, the neutrophils were showing cytoplasmic vacuoles with a shadowy appearance (Fig. 2e) which raised a suspicion of negative staining inside the vacuoles and prompted us to do a Z-N stain on the peripheral blood. It was positive for acid-fast bacilli within the neutrophils (Fig. 2f) suggestive of tubercular bacterium. In view of the granulocytic phagocytic activity, further biochemical tests were done to rule out the possibility of haemophagocytic lymphohistiocytosis (HLH). He was found to have hyperferritinaemia (> 95,000 ng/mL), hypertriglyceridaemia (747 mg/dL), hypofibrinogenaemia (86.5 mg/dL) with bicytopaenia (anaemia and thrombocytopenia), fever, and splenomegaly which fulfilled 5 out of 8 clinical and laboratory criteria for HLH. A flow cytometry lymphocyte subset analysis of T, B, and NK cells did not elicit any quantitative cellular deficiencies. A bone marrow study could not be performed as his clinical condition worsened rapidly with renal and hepatic failure and he succumbed before any specific therapy could be started.

a Neutrophils show cytoplasmic vacuoles and the phagocytosis of the apoptotic cellular debris (pointed by the arrow) (Leishman Giemsa stain, × 1000). b Neutrophils with the Chediak-Higashi-like inclusion (pointed by the arrow) (Leishman Giemsa stain, × 1000). c Shows an eosinophil and a neutrophil with blue-green inclusions (pointed by the arrow) (Leishman Giemsa stain, × 1000). d The background apoptotic and necrotic “cellular debris” (pointed by the arrow) that led to spuriously elevated platelet counts (Leishman Giemsa stain, × 1000). e Neutrophils with vacuoles showing shadowy appearance (pointed by the arrow) (Leishman Giemsa stain, × 1000). f Z-N stain showing acid-fast bacilli (pointed by the arrow) with inset demonstrates intracellular localization of AFB (× 1000)
All the peripheral blood samples were analysed on Sysmex XN 1000 automated CBC analyser. When the analyser-based expression of WBC scatter plot was compared with normal WBC expression for any specific variation, it was found to mimic the expression of haematolymphoid neoplasm especially juvenile myelomonocytic leukaemia. The analyser-based judgement was showing interpretive program (IP) message or flagging of WBC abnormal scatter gram, leukocytosis, neutrophilia, eosinophilia, monocytois, lymphocytosis, basophilia, NRBC, immature granulocytes, and blasts or abnormal lymphocytes. The cell population data (CPD) available in the cell counter as research parameter were also analysed. The neutrophils (NE) were showing higher cellular complexity with wider dispersion as reflected objectively from the cytometry channel values (side scatter NE-SSC 147.5, width of SSC dispersion NE-WX 536, side fluorescence NE-SFL 80.3, width of SFL dispersion NE-WY 1495, forward scatter NE-FSC 88.0, and width of FSC dispersion NE-WZ 149). Similarly, wider variations were noted in lymphocytes (LY) and monocytes (MO) population data (lymphocyte side scatter LY-X 83.0, lymphocyte SSC dispersion LY-WX 590, lymphocyte side fluorescence LY-Y 64.0, lymphocyte fluorescence dispersion LY-WY 1391, lymphocyte size LY-Z 61.1, lymphocyte size dispersion LY-WZ 704, monocyte side scatter MO-X 106.8, monocyte side scatter dispersion MO-WX 449, monocyte side fluorescence MO-Y 104.6, monocyte fluorescence dispersion MO-WY 1119, monocyte size MO-Z 69.7, and monocyte size dispersion MO-WZ 703). These wider variations are attributed to cytoplasmic vacuolations, phagocytosis, blue-green inclusions, Chediak-Higashi-like inclusions, and intracellular tubercular bacilli. The corresponding scatter plots were as depicted in Figs. 3 and 4.

a The normal SSC vs. SFL scatter plot of WDF channel in the Sysmex XN analyser. It shows the normal distribution of lymphocytes (pink), monocytes (green), neutrophils (light blue), eosinophils (red), and debris (dark blue). b Scatter plot of SSC vs. SFL in WDF channel of our case. It shows all grey-coloured events which indicate that the cellular events are outside the areas as depicted in normal reference scatter plot. The debris area also shows very high numbers of events indicative of increased cellular debris being spuriously counted as platelets giving rise to pseudo-thrombocytosis

a Normal FSC vs. SFL scatter plot in WNR channel in Sysmex XN analyser. b The FSC vs. SFL scatter plot in WNR channel of our case shows increased nucleated RBC (pink), increased debris (dark blue), and basophilia (yellow) with the leukocytosis (light blue)
Discussion
It is nearly a century since the BCG vaccine has been developed by French bacteriologists Albert Calmette and Camille Guérin. Currently, it is one of the most widely used vaccines which are being routinely administered at birth as part of World Health Organization global expanded immunisation program. It is considered to have an excellent safety profile. However complications can occur with one in 1000 developing serious local reactions but severe disseminated diseases are rare with a reported incidence of less than one in a million cases. A review by Lotte et al. identified 60 cases of disseminated BCG infection in literature from 1921 to 1982 with a high mortality rate of 50% [5]. Similarly, Elizabeth et al. identified additional 28 cases between 1980 and 1997 [6]. A meta-analysis by Saied Mostaan et al. reported 44 cases of disseminated BCG infection between 1997 and 2016 [7]. The primary or secondary immunodeficiency diseases are at risk of severe and life-threatening BCG-osis with an overall mortality rate of 30% [8]. A number of primary immunodeficiency diseases (PIDs) like severe combined immunodeficiency (SCID), chronic granulomatous disease (CGD), and Mendelian susceptibility to mycobacterial diseases have been reported to have increased susceptibility to severe mycobacterial infections following vaccination with BCG [9].
The BCG-osis commonly affects male with onset of adverse reactions within 5 months. Consanguinity is evident in some cases indicative of the role of genetic predisposition or increased susceptibility to primary immunodeficiency diseases. Fever is the most common manifestation followed by skin lesions, lymphadenopathy, osteomyelits, hepatosplenomegaly respectively [8]. There is no established definition or diagnostic criteria for disseminated BCG infection or BCG-osis but the working definition of Elizabeth et al. will be of help for diagnosing cases. It needs at least BCG to be cultured and identified by biochemical methods; evidence of dissemination by blood or bone marrow culture, or evidence of infection at two or more anatomic sites beyond the region of vaccination; and a systemic syndrome compatible with mycobacterial disease, i.e., typical manifestations of fever, weight loss, anaemia, and death [6]. In our case, the presence of persistent fever, failure to thrive, hepatosplenomegaly, erythematous skin lesions, lymphadenopathy with ulceration, and sparse depigmented hair raised the initial possibilities of bacterial infection, congenital or disseminated tuberculosis, haematological malignancies, or metastatic malignancies. Initial investigations did not reveal any specific evidence of bacterial sepsis as the blood, CSF, and urine aerobic culture were negative. A peripheral blood leukoerythroblastic blood picture and absolute monocytosis raised the differential possibility of juvenile myelomonocytic leukaemia (JMML) but the Haemoglobin F levels of 1.4% did not support it. The presence of acid-fast bacilli in gastric aspirate, axillary lymphadenopathy with ulceration on the same side of BCG inoculation, and systemic manifestations taken together were suggestive of the diagnosis of BCG-osis. It was further supported by positive mycobacterial culture.
An underlying immunodeficiency disorder was evaluated. The HIV screening was negative. The lymphocyte subset analysis by flow cytometry did not show any cellular deficiency of T, B, or natural killer cells. There was no hypo-gammaglobulinaemia. But possibility of coexisting immunodeficiency cannot be ruled out in our case in view of the family history of consanguinity. A peripheral blood with pseudo-Chediak-Higashi-like inclusions further supports such a possibility. Chediak-Higashi syndrome is caused by the mutation of CHS1/LYST gene impairing the normal function of the lysosomal trafficking regulator protein. It is characterised by repeated and persistent infection starting from infancy or childhood, oculocutaneous albinism, coagulopathy, and varying neurological impairment. The presence of cytoplasmic giant granules are readily appreciated in leukocytes in most of the cases [10, 11]. In our case, there was leukocytosis contrary to classical findings of pancytopenia in Chediak-Higashi syndrome and absence of oculocutaneous albinism further ruled out such a possibility. Hence, we proposed these giant cytoplasmic granules mimicking Chediak-Higashi as pseudo-Chediak-Higashi granules associated with this specific disorder of unknown aetiology.
Our case also presented with haemophagocytic lymphohistiocytosis. The neutrophils with phagocytosis of apoptotic debris formed the basis to investigate the possibility of haemophagocytic syndrome. There is lack of any such specific evidence of haemophagocytosis with BCG-osis but few cases of disseminated tuberculosis with haemophagocytosis in infants have been reported in literature [12]. Karaca et al. have reported a case of disseminated BCG infection with hyperferritinaemia with a novel NEMO mutation [13].
The microorganisms are phagocytosed and killed by the neutrophils and monocytes in the peripheral blood but Mycobacterium is relatively protected and survives phagocytosis [14]. The demonstration of acid-fast bacilli within the neutrophils by Z-N stain in our case is direct evidence to it. The presence of cytoplasmic shadowy vacuoles in neutrophils can be a clue to such a possibility. There are reports of Mycobacterium within macrophages seen as negative images in cytology smears especially from patients with acquired immunodeficiency syndrome [15]. In addition, the neutrophils were showing blue-green cytoplasmic inclusions. Green cytoplasmic inclusions are a rare finding with a total of 41 cases reported in the literature. Clinically, the inclusions are associated with elevated liver enzymes, hepatic failure and a high early mortality rate [16]. Similarly, our case also presented with hepatic failure and an early fatal outcome. Other unique feature was presence of cellular debris in the peripheral blood giving rise to spurious thrombocytosis by the cell counter. This indicates the importance of smear review to rule out the possibility of pseudo-thrombocytosis due to increase in circulating cellular debris. Hence, the peripheral blood must be carefully evaluated to screen for morphologically significant findings which may have diagnostic and prognostic importance. In addition, screening scatter plots, histograms, and research cell population data of the automated cell counters can provide further diagnostic clue. Hence, we have recorded the haematology analyser-based expression of the peripheral blood tubercular bacteraemia which can be of much help for future studies.
Our investigation is limited by absence of evaluation for immunoglobulin (IgG, IgM, and IgA) levels and molecular studies to identify the presence of any specific primary immunodeficiency disorders. An autopsy could have yielded more information in our case.
Conclusion
Infants with an ipsilateral lymphadenopathy on the side of BCG vaccine inoculation with systemic manifestation should be considered for a possibility of BCG-osis with investigation to rule out any underlying immunodeficiency. A possibility developing HLH need to be ruled out as it may be life-saving. The morphological clue in the peripheral blood and haematology analyser-based scatter plot should also be correlated for additional evidence.
References
SAGE Working Group on BCG Vaccines and WHO Secretariat (2017) Report on BCG vaccine use for protection against mycobacterial infections including tuberculosis, leprosy, and other nontuberculous mycobacterial (NTM) infections. http://www.who.int/immunization/sage/meetings/2017/october/1_BCG_report_revised_version_online.pdf. Accessed: 15-Apr-2018
Hesseling AC, Johnson LF, Jaspan H, Cotton MF et al (2009) Disseminated bacille Calmette–Guérin disease in HIV-infected South African infants. Bull World Health Organ 87:505–511. https://doi.org/10.2471/BLT.08.055657
Lotte A et al (1988) Second IUATLD study on complications induced by intradermal BCG-vaccination. Bull Int Union Tuberc Lung Dis 63(2):47–59
Padhi S, Ravichandran K, Sahoo J, Varghese RG, Basheer A (2015) Hemophagocytic lymphohistiocytosis: an unusual complication in disseminated Mycobacterium tuberculosis. Lung India 32(6):593–601
Lotte A, Wasz-Hockert O, Poisson N, Dumitrescu N, Verron M, Couvet E (1984) BCG complications: estimates of the risks among vaccinated subjects and statistical analysis of their main characteristics. Adv Tuberc Res 21:107–193
Talbot EA, Perkins MD, Silva SFM, Frothingham R (1997) Disseminated Bacille Calmette-Guérin disease after vaccination: case report and review. Clin Infect Dis 24(6):1139–1146
Mostaan S, Yazdanpanah B, Moukhah R, Hozouri HR, Rostami M, Khorashadizadeh M et al (2016) Adverse effects of BCG vaccine 1173 P2 in Iran: a meta-analysis. Adv Biomed Res 5:99
Shahmohammadi S, Saffar MJ, Rezai MS (2014) BCG-osis after BCG vaccination in immunocompromised children: case series and review. J Pediatr Rev 2(1):62–74
Norouzi S, Aghamohammadi A, Mamishi S, Rosenzweig SD, Rezaei N (2012) Bacillus Calmette Guérin (BCG) complications associated with primary immunodeficiency diseases. J Inf Secur 64(6):543–554
Kaplan J, De Domenico I, Ward DM (2008) Chediak-Higashi syndrome. Curr Opin Hematol 15(1):22–29
Roy A, Kar R, Basu D, Srivani S, Badhe BA (2011) Clinico-hematological profile of Chediak Higashi syndrome: experience from a tertiary care center in south India. Indian J Pathol Microbiol 54(3):547–551
Deshpande A, Nayar PS, Pradhan AM, Manchanda RV (2010) Miliary tuberculosis with hemophagocytosis in a two months old infant. Indian J Hematol Blood Transfus 26(3):115–117
NE K et al (2016) Disseminated BCG infectious disease and hyperferritinemia in a patient with a novel NEMO mutation. J Investig Allergol Clin Immunol 26(4):268–271
Dale DC, Boxer L, Liles WC (2008) The phagocytes: neutrophils and monocytes. Blood 112(4):935–945
Maygarden SJ, Flanders EL (1989) Mycobacteria can be seen as ‘negative images’ in cytology smears from patients with acquired immunodeficiency syndrome. Mod Pathol 2(3):239–243
Yang J, Gabali A (2018) Green neutrophilic inclusions: current understanding and review of literature. Curr Opin Hematol 25(1):3–6
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Bhola, R.K., Sarangi, R., Dey, P. et al. Disseminated BCG-osis with haemophagocytosis, tubercular bacteraemia, and unusual haematological findings with its haematology analyser-based expression. J Hematopathol 11, 87–92 (2018). https://doi.org/10.1007/s12308-018-0327-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-018-0327-1