
Abstract
Overexpression of p53 tumor suppressor protein in malignant cells induces cell cycle arrest, or alternatively, apoptosis thereby indicating that additional factors may contribute to the p53-mediated outcome. Comparison of the experimental protocols revealed that the construct encoding wild-type (wt) p53 was expressed in cells of different origin. Therefore, we decided to determine whether the intrinsic cellular program of primary cells of the same genetic background could have any effect on the oncogenic potential of mutated c-Ha-RAS and TP53. Primary rat cells (RECs) isolated from rat embryos of different age: at 13.5 gd (y) and 15.5 gd (o), were used for transfection. Immortalized rat cell clones overexpressing temperature-sensitive (ts) p53135val mutant and transformed cell clones after co-transfection with oncogenic c-Ha-Ras, were generated. The ts p53135Val mutant, switching between wt and mutant conformation, offers the possibility to study the role of p53 in cell cycle control in a model of malignant transformation in cells with the same genetic background. Surprisingly, the kinetics of cell proliferation at non-permissive temperature and that of cell cycle arrest at 32°C strongly differed between cell clones established from yRECs and oRECs. Furthermore, the kinetics of the re-enter of G1-arrested cells in the active cell cycle strongly differed between distinct cell clones. Finally, the susceptibility of immortalized and transformed cells to the pharmacological inhibitors of cyclin-dependent kinases (CDKs) considerably differed. Our results clearly show that overexpression of genes such as mutated TP53 and oncogenic c-Ha-RAS is not able to fully override the intrinsic cellular programme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The decision of a cell to stop cell cycle progression and to initiate the repair of (mildly) damaged DNA, or to induce apoptosis as a consequence of rather severely damaged DNA, bears fundamental implications on the future development, well-being, and fate of the whole organism. In case repair does not function properly or the induction of apoptosis is impaired, neoplastic transformations arising from damaged DNA, might culminate in the death of the whole organism. Consequently, in the case of apoptosis a single cell is sacrificed to facilitate the survival of the being. Therefore, an extremely sophisticated cellular network protects the integrity of the genome and induces the necessary steps once this integrity is disrupted.
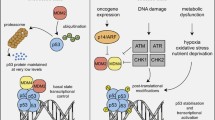
At the interface between the incoming intra- and extracellular signals and the downstream induction and execution of cell cycle arrest and apoptosis, higher eukaryotic cells have a molecule of paramount importance: the p53 tumor suppressor protein. In most cases of cellular damage p53 is involved in the decision to trigger cell cycle arrest or apoptosis. Additionally, p53 is involved in all 5 major pathways for DNA repair [2, 20, 26, 35]. The fact that p53 is inactivated in a wide variety of tumors, underscores its importance and makes it an outstanding candidate for cancer therapy [3, 34].
p53 transmits its signals through transactivation of target genes but also through direct binding to other proteins. In the cell, p53 levels rise as a result of certain stress stimuli but are otherwise kept low due to the action of a negative feedback loop with MDM2. If the action of p53 is needed, a variety of proteins disrupt the interaction between p53 and MDM2 [29]. A protein strongly contributing to the stability of p53 is poly(ADP-ribosyl) polymerase-1 (PARP-1) [38, 42, 43], a protein that enzymatically modifies p53 [19, 41] thereby preventing its nuclear export [19, 39] by impeding the binding to CRM1 [19]. A protein that retains p53 in the cytoplasm preventing its nuclear functions, is mortalin, a member of the heat shock protein 70 (HSP70) family. Mortalin binds p53 [31] and inhibits its pro-apoptotic functions what leads to increased tumor development [31, 37].
The constitutive overexression of p53 in cells or animals is not feasible because this would trigger apoptosis or at least cell cycle arrest, making a functional study of the proteins’ features impossible. Fortunately, a temperature-sensitive (ts) mutant of p53 that displays wt properties at 32˚C but mutant character at elevated temperature [25], can be used to perform experiments aimed to elucidate its functions. This ts mutant demonstrates clear properties of mutant p53 at 39°C. At 37°C the cells also behave like mutant cells although a small portion of p53 protein is in wt conformation. However, mutated p53 protein localized in the cytoplasm impedes the action of the wt protein. Thereby, the conformation and activity of p53 can be changed at will by simply growing the cells at 37 or 39˚C.
The decision of p53 to trigger cell cycle arrest or apoptosis depends on the severity of the damage and is also regulated on the transactivational level by the use of p53 responsive elements to which the protein has different binding affinity [16]. In general, p53 binds to targets mediating cell cycle arrest with a higher affinity than to those which induce apoptosis [16]. A recent publication also showed that p53 is capable of inducing anti-apoptotic targets [17], adding further complexity to the functions and activities of the tumor suppressor protein.
Also the Ras proteins are important for tumor development. In their active form they reside in the cytoplasmatic membrane and transmit signals from growth factor stimulation and downstream targets involve Raf-1 and PI3-kinase. Gain of function mutations lead to a constitutively active Ras protein that sustains growth-promoting signals, irrespective of extracellular stimulation, resulting in uncontrolled proliferation. For its proper anchoring in the cytoplasmic membrane and activity, Ras has to be isoprenylated by farnesyl protein transferases (FPTases) or/and geranylgeranyl protein transferases. Therefore, inhibitors of farnesylation have been used for treatment of cancers with constitutively activated RAS. Interestingly, tumor cells with constitutively activated RAS are rendered prone to treatment with pharmacological inhibitors of cyclin-dependent kinases (CDKs) like roscovitine (ROSC) and olomoucine (OLO) when they are pre-treated with FTIs [45].
To test the responsiveness of cells overexpressing c-Ha-Ras and ts p53, we used primary rat embryonal cells (RECs) isolated at 13.5 or 15.5 gestation days. Importantly, the age of the embryos seems to have an effect on the properties of the cells [30]. In the present work we investigated the effect of the cellular microenvironment in young vs old RECs in response to combined treatment with FPTase inhibitors and CDK inhibitors.
Material and Methods
Plasmids
pLTRp53cGval135, comprising a chimera of mouse p53 cDNA and genomic DNA (generous gift of Dr. M. Oren), has been previously referred to as pLTRp53cG [6]. It encodes a mutant protein harboring a substitution from alanine to valine at the amino acid in position 135. The plasmids pVV2, bearing the neomycin resistance sequence, and pVEJB coding for a mutated human c-Ha-Ras gene cloned into pVVJ were used.
Cell Clones
The transformed rat cell clones were established as previously described in detail [40] using primary Fisher rat embryo cells (RECs). RECs were obtained from embryos isolated at 13.5 (y) and 15.5 (o) gestation days. Cells were grown at basal temperature (37°C) in DMEM supplemented with 10% FCS in an atmosphere of 7.5% CO2. For experiments dealing with a change of the conformational state of p53 protein, cells grown at basal temperature, were shifted to 32°C for indicated periods of time.
Drugs
Olomoucine (OLO) and roscovitine (ROSC) were prepared as 50 mM stock solution in DMSO according to the published procedure [14]. Aliquots of the stock solution were stored until use at -20°C. Furthermore, L-744,832 [(2 S)-2-[[(2 S)-2-[(2 S,3 S)-2-[(2R)-2-amino-3-mercaptopropyl]amino]-3-methylpentyl]oxy]-1-oxo-3-phenylpropyl]amino]-4-(methylsulfonyl)-butanoic acid 1-methylethyl ester ] an inhibitor of protein farnesyltransferase (FTI) from Alexis Biochemicals (Lausen, Switzerland) was used. The stock solution of L-774,832 was prepared in DMSO. Aliquots of stock solutions were protected from light and stored until use at -20°C.
Cell Treatment
After plating, cells were cultivated at a basal temperature of 37°C for 24 h. Then drugs were added to a final concentration as indicated. Cells were incubated continuously for 24 h or 48 h, or alternatively, after 24 h treatment medium was changed and cells were post-incubated (p. i.) in a drug-free medium for a further 24 h or 48 h. In some experiments cells were shifted to 32°C and kept there for at least 24 h prior to the onset of treatment to allow p53 to adopt wt conformation.
Determination of Population Doubling Time
To determine the kinetics of the proliferation of distinct cell clones, cells were plated into PD of 6 cm diameter. For each time point two PDs were used. Immortalized cells were plated at a medium density (2 × 105) and transformed cells at a lower density (0.5 × 105/PD). Cells were cultivated at a basal temperature for 5 days. PDs were collected in 12 h intervals, suspended in a defined volume of medium and were counted in a cell counter (CASY). Cell number was determined in at least two distinct aliquots of cell suspension collected from each PD.
Determination of the Number of Viable Cells
Proliferation of immortalized and transformed control rat cells and their sensitivity to increasing concentrations of the CDK inhibitor ROSC were determined by the CellTiter-GloTM Luminescent Cell Viability Assay (Promega Corporation, Madison, WI). As described recently in more detail [44], the CellTiter-GloTM Luminescent Cell Viability Assay, generating a luminescent signal, is based on quantification of the cellular ATP levels. Tests were performed at least in quadruplicates. Luminescence was measured in the Wallac 1420 Victor, a microplate luminescence reader. Each point represents the mean ±SD (bars) of replicates from at least four experiments.
Determination of Caspase-3/7 Activity
The activity of both caspases was determined using the APO-ONE Homogenous Caspase-3/7 Assay (Promega, Madison, WI) which uses the caspase-3/7 substrate rhodamine 110, bis-(N-CBZ-L-aspartyl-L-glutamyl-L-valyl-L-aspartic acid amide) (Z-DEVD-R100) as described previously [44]. Briefly, rat cells were plated in 96-well microtiter plates. One day after plating the cells were exposed for 24 h to increasing drug concentrations. Thereafter, culture supernatant was transferred into another microtiter plate to separately determine the caspase activity in cells and in culture medium. Then an equal volume of caspase substrate was added and samples were incubated at 37°C for different periods of time to assess the best signal-to-background ratio. The fluorescence was measured at 485 nm. Luminescence and fluorescence were measured in the Wallac 1420 Victor, a microplate luminescence reader. Each point represents the mean ± SD (bars) of replicates from at least three experiments.
Measurement of the DNA Content of Single Cells by Flow Cytometry
The measurement of DNA content was performed by flow cytometric analysis based on a slightly modified method [38] described previously [36]. The cells were detached from substratum by trypsinization, and then all cells were harvested by centrifugation and washed in PBS. Aliquots of 1 × 106 cells were used for further analysis. Cells were stained with propidium iodide (PI) as described, previously [39]. Fluorescence was measured using the Becton Dickinson FACScan after at least 2 h incubation of the cells at +4°C in the dark.
Results
Differential Proliferation Rate of y and o Immortalized Rat Cells
In the first step the proliferation rate of primary rat cells and four studied cell clones were determined. Cells plated in the defined cell density were cultivated for 5 days at a basal temperature. Cell numbers were determined in 12 h intervals by two different methods. First, cells were counted using an automatic cell counter and in parallel numbers of living cells were determined by the CellTiter-GloTM Luminescent Cell Viability Assay (Promega Corporation, Madison, WI). As described recently in more detail [44], the CellTiter-GloTM Luminescent Cell Viability Assay, generating a luminescent signal, is based on quantification of the cellular ATP levels. Both methods gave comparable results within the first 3 days; later CellTitert-Glo underestimated the numbers of transformed cells. The primary cells isolated from rat embryos (RECs) at gestation day 13.5 (y) grew much slower than those isolated at day 15.5 (o) [30]. The population doubling time (PDT) calculated from growth curves for the oRECs was approximately 2-fold shorter than that for yRECs (Table 1).
RECs transfected with ts p53135Val mutant alone, or simultaneously with human c-Ha-RAS, generated immortalized and transformed cells, respectively. As described previously [30], the phenotype of immortalized cells resembled that of primary cells. However, in contrast to RECs, cells expressing ts p53135Val got over the Hayflick limit and did not undergo senescence. Cotransfection with c-Ha-RAS resulted in a clear change of cell morphology to spindle-shaped [30] and conferred the generated cell lines a high mitotic potential. The features of transformed cells were also functionally proved. After subcutaneous injection of cells overexpressing p53135Val +c-Ha-Ras into rats large tumors appeared within approximately 2 weeks [30].
As shown in Fig. 1, the transformed cells divided very rapidly. Interestingly, the immortalized and transformed cell lines originating from oRECs (clone 602/534 and 173/1022), divided at 37°C much more rapidly than those from yRECs (clone 402/534 and 189/111). The population doubling time (PDT) was calculated for each cell clone from the growth curves. As depicted in Table 1, even immortalization of yRECs with ts p53135Val mutant (clone 402/534) did not confer them high mitotic potential at non-permissive temperature. On the other hand, the proliferative potential of their counterparts generated from oRECs was markedly higher. Interestingly, the same trend was observed in transformed cell lines after co-transfection with c-Ha-RAS. However, during the time period between 24 h and 48 h after cell plating, the transformed cells did not gain the full dividing capacity. The difference in the proliferation rate between transformed y and o cell clones became evident 48 h after cell plating (Fig. 1).

Kinetics of proliferation of immortalized and transformed rat cells. Immortalized (402/534 and 602/534) and transformed (189/111 and 173/1022) cell clones established in RECs from embryos at 13.5 (y) and 15.5 (o) gestation days were examined. The growth curves of immortalized and transformed RECs from young (402/534 and 189/111) and old (602/534 and 173/1022) embryos at basal temperature are shown. Cells were counted in a cell counter (CASY). Each point represents the mean of four cell aliquots ± SD. Transformed cells grow faster than primary cells. The cells originating from older embryos always grow faster than their counterparts from young embryos. Population doubling time (PDT) for each cell line is shown in Table 1. 402/534 - yRECs p53135Val; 602/534 - oRECs p53135Val; 189/111 - yRECs p53135Val + c-Ha-Ras; 172/1022 - oRECs p53135Val + c-Ha-Ras
Kinetics of wt p53-Mediated Cell Cycle Arrest Differs Between Cell Clones Generated in y and o Embryonal Rat Cells
In accordance with previous reports, in cells overexpressing ts mutant p53135Val, the protein switches conformation after temperature shift to 32°C and as a consequence, cells start to accumulate in G1 phase of the cell cycle (Fig. 2). The induction of cell cycle arrest after temperature shift to 32°C was observed solely in cells expressing ts mutant p53135Val but not in cells overexpressing c-myc + c-Ha-Ras (our unpublished data) and was associated with the translocation of p53 protein from the cytosol to the nucleus [30, 37, 41]. Moreover, primary yRECs and oRECs lacking the ts mutant and expressing endogenous p53 at low concentrations failed to accumulate in G1 phase after maintenance at 32°C [30]. These observations substantiate the assumption that the temperature-dependent block of cell proliferation and of the cell cycle progression at permissive temperature is attributable to ts p53 mutant and evidence that the experimental system functions properly.

Intrinsic features of RECs determine the p53-mediated cell cycle regulation. DNA profile obtained from one representative experiment. Young immortalized (first horizontal row), old immortalized (second horizontal row), young transformed (third horizontal row) and old transformed cells (fourth horizontal row) were cultivated at 37˚C for 24 h and then shifted to 32˚C for 24 h. DNA concentration in single cells was determined by flow cytometric analysis of PI-stained cells. DNA histograms were prepared using the CellQuest evaluation program (upper panel). The frequency of diploid cells in the distinct cell cycle phases was determined using the ModFit evaluation program (lower panel)
After maintenance for 24 h at permissive temperature, the population of S-phase cells was strongly reduced in all four cell lines. However, the frequency of the G2/M population varied between them. The comparison of the time course of the cell cycle changes revealed considerable differences in the kinetics of the cell cycle arrest at permissive temperature as shown in Fig. 3. The immortalized 402/534 cells were almost completely arrested in G1 after 24 h at 32°C, whereas in 602/534 cells only S-phase, but not G2 phase was diminished (Fig. 3, upper panel). In contrast, transformed cells (clone 173/1022) generated from oRECs, showed a stronger response to the temperature shift.

Kinetics of the cell cycle arrest in the permissive (32°C) temperature. The FACS analyses show the cell cycle distribution of immortalized, and transformed cells originating from young (left panels) and old (right panels) RECs at 32 and 37˚C. oRECs more efficiently evade cell cycle arrest than yRECs in all groups. As expected, immortalized cells show stronger growth than primary cells and transformed cells exhibit the strongest growth. The frequency of diploid cells in the distinct cell cycle phases was determined using the ModFit evaluation program. The values represent the means of three independent experiments ± SD (bars)
G1-arrested, Transformed Rat Cells Re-enter more Rapidly the Active Cell Cycle than their Immortalized Counterparts
In the next series of experiments we addressed the question whether the endogenous features of primary cells used for establishment of cell lines might display any effect on the recovery of G1-synchronized cells in the active cell cycle. We maintained all cell clones for 24 h at permissive temperature and then shifted them back to the basal temperature. As depicted in Fig. 4, transformed cells entered the active cell cycle more rapidly than the immortalized cells. Surprisingly, the kinetics of cell cycle recovery strongly differed between cell lines derived from y and o RECs. In the latter a pronounced increase of S-phase cells was observed 6 h after elevation of temperature and after a further 6 h the ratio of DNA-replicating cells was approximately 70%. Moreover, maintenance of examined rat cells at permissive temperature slightly increased the ratio of sub-G1 cells indicating that this subset of cells represents apoptotic cells. To check it, the activity of caspase-3/7 was determined. A moderate elevation of the activity of effector caspases was observed in 402/534 and 189/111 cells (data not shown) confirming the assumption that at permissive temperature wt p53 may induce apoptosis.

Temperature-dependent kinetics of proliferation of primary, immortalized, and transformed rat cells. RECs were isolated from embryos at 13.5 (y) and 15.5 (o) gestation days. The growth curves of primary, immortalized, and transformed RECs from young (left vertical row) and old (right vertical row) embryos at three different temperatures are shown. Immortalized cells grow faster than primary cells and transformed cells grow fastest. The cells originating from older embryos always grow faster than their counterparts from young embryos. The values represent the means of three independent experiments ± SD (bars)
The Pharmacological Inhibitors of CDKs Stronger Affect Transformed Rat Cells Established from Primary Cells Isolated at 13.5 gd than Cells Isolated at 15.5 gd
To determine the effect of both examined CDK inhibitors on the proliferation of exponentially growing transformed rat cells, the cells were continuously exposed to the drugs for 24 h or 48 h. Then the cell number was determined using the CellTiterLumiGlo viability assay immediately upon termination of the treatment, or alternatively the medium was changed, cells were post-incubated in a drug-free medium for further 1 or 2 days, and then the assay was performed. OLO is a weaker inhibitor of CDKs than ROSC [14] and therefore we used it at a higher dosage. As expected, ROSC stronger reduced the number of living cells than OLO. Moreover, transformed cells established from primary rat cells isolated at 13.5 gd (189/111 cells) were more sensitive to the inhibition of CDKs than their counterparts generated from 15.5 gd RECs (173/1022) (Fig. 5). Exposure of 189/111 cells to ROSC at a final concentration of 20 µM reduced the number of living cells by approximately 30% and the number of 173/1022 cells by approximately 15%. The anti-proliferative effect of ROSC at higher dosage was very highly significant in both cell lines after treatment for 24 h (Fig. 5) and 48 h (data not shown).

The pharmacological inhibitors of CDKs stronger affect transformed rat cells established from primary cells isolated at 13.5 gd than from cells isolated at 15.5 gd. Transformed cells were plated into 96 well microtiter plates (two plates for each condition). One day after plating, cells were exposed to drugs for 24 h or for 48 h (not shown). Thereafter, the number of viable cells was determined using CellTiterGlo. Tests were performed at least in quadruplicate. Luminescence was measured in the Wallac 1420 Victor, a multilabel, multitask plate counter. Each point represents the mean ± SD (bars) of replicates from three independent experiments. Statistical analysis was performed using GraphPad Prism and significance levels were evaluated using T test
Inhibition of c-Ha-Ras Processing Sensitizes Transformed Rat Cells Established from oRECs to CDK Inhibitors
Further, we addressed the question whether the activity status of overexpressed oncogenic c-Ha-Ras might have any effect on the susceptibility of transformed rat cells to tested CDKs inhibitors. To gain full biological activity, Ras proteins after de novo synthesis have to be stepwise modified. Isoprenylation, catalyzed by farnesyl protein transferase (FPTase), is the first reaction in this series of events. Both cell lines were treated for 24 h with L-744,832, a pharmacological inhibitor of FPTase (FTI) alone or in combination with OLO or ROSC. Then the number of living cells was determined immediately or alternatively, medium was changed and cells were post-incubated for 24 h in a drug-free medium or with FTI. The inhibition of isoprenylation had a stronger anti-proliferative effect on 173/1022 than on 189/111 cells (Fig. 6). Addition of FTI to ROSC enhanced its inhibitory effect on 173/1022 cells. The strongest reduction of the number of viable 173/1022 cells occurred after post-incubation for 24 h in the presence of FTI (Fig. 6).

Inhibition of c-Ha-Ras processing sensitizes transformed rat cells established from oRECs to CDK inhibitors. Transformed cells were plated into 96 well microtiter plates (two plates for each condition). One day after plating, cells were exposed to indicated drugs for 24 h. Thereafter, the number of viable cells was determined in the first microtiter plate. In the second microtiter plate medium was changed (MC) and cells were post-incubated (p.i.) for a further 24 h in a drug-free medium or with FTI. The measurement of the number of viable cells immediately after treatment for 24 h provided information on the direct cytotoxic effect of the drug. On the other hand, post-incubation of cells treated for 24 h, for another 48 h in a drug-free medium, allowed the evaluation of the long-term effects of the treatment. Tests were performed at least in quadruplicate. Luminescence was measured in the Wallac 1420 Victor, a multilabel, multitask plate counter. Each point represents the mean ± SD (bars) of replicates from three experiments. Statistical analysis was performed using GraphPad Prism and significance levels were evaluated using T test
Taken together, our above results show that immortalized and transformed cell lines established from primary cells isolated from older embryos (15.5 gd) had a proliferation advantage over their counterparts isolated from younger embryos (13.5 gd) associated with less susceptibility to therapy. It seems that c-Ha-Ras, when overexpressed in oRECs, contributes to their lower susceptibility to synthetic CDK inhibitors.
Discussion
For investigations concerning tumor development and also the treatment of cancer, the analysis of properties from tumor suppressor proteins as well as from oncogenes is of paramount importance. Since the TP53 and RAS genes are two of the most frequetly affected targets during neoplastic transformation in a wide variety of cells and tissues [11, 13], we focused our research presented here, on these two molecules. The RAS proto-oncogene is often mutated, leading to a constitutively active form and p53 is usually inactivated or expressed as a dominant negative protein in tumors.
Most importantly, inactivated TP53 and mutated c-Ha-RAS act synergistically in making cells vulnerable to chemically induced carcinogenesis in vitro and also in vivo [47, 48]. The ts p53 used in our work was shown to synergistically induce malignant transformation together with c-Ha-Ras in primary RECs [12]. Hemizygosity in p53 leads to clear signs of haploinsufficiency [10, 15] and germ line mutations in humans are known as Li-Fraumeni syndrome [23] leading to multiple cancers with poor prognosis [7]. The synergistic action of mutated TP53 and c-Ha-RAS in tumor development and progression [32, 47] is not surprising, considering that p53 protein usually arrests the cell cycle of damaged cells or induces apoptosis, and Ras is able to transmit extracellular, growth-promoting signals via the Ras/Raf/MEK/ERK pathway [21].
Several isoforms of the two other members of the p53 family, p63 [46] and p73 [4], have overlapping functions with p53 and some isoforms have rather opposing functions. Therefore, these proteins are important for fine-tuning and play additional roles in early development, but they are not able to take over the functions of inactivated p53.
In the present work we used primary, immortalized (ts p53), and transformed (ts p53 and c-Ha-Ras) RECs from young (13.5 gd) and old (15.5 gd) embryos to compare their growth potential and their susceptibility to treatment with FPTase inhibitors and CDK inhibitors. At the basal temperature (37˚C; p53 inactive) the immortalized and transformed cell lines originating from oRECs (clones 602/534 and 173/1022, respectively) showed a clearly elevated growth potential as compared to their counterparts from yRECs (402/534 and 189/111, respectively). Not surprisingly, transformed cells in both cases grew faster than immortalized cells from the same kind of embryos (y vs o). Apparently, epigenetic changes take place between 13.5 and 15.5 gestation days, leading to an elevated potential of cells from older embryos to overcome growth arrest.
Next we tested the effect of the CDK inhibitors roscovitine and olomoucine on transformed cells from young and old embryos. The transformed cells from young embryos were more sensitive to treatment with CDK inhibitors than their counterparts from older embryos. Most importantly, following prior treatment with an FPTase inhibitor that inactivates c-Ha-Ras, also transformed cells from older embryos were strongly susceptible to the growth-inhibiting effect of CDK inhibitors. These results show, that c-Ha-Ras contributes to the partial resistance of transformed cells from oRECs to the action of CDK inhibitors. A thorough scrutiny of the exact mechanistic background for the differences in the behaviour of the mentioned cell types should shed additional light on the cellular basis for the described effects.
In distinct stages of embryonic development tissue homeostasis is modulated by a balance between proliferation and programmed cell death. A temporally and spatially regulated apoptosis is essential for differentiation and maturation of different tissues and plays an important role, especially in neurogenesis. The increase of apoptotic events occurs in mid stages of embryonic development. Analyses of rat fetuses from the biologically most interesting stages revealed differences in the expression of some important proteins including CDK5 [5, 27] or alpha-fetoprotein [24].
The epigenetic changes between 13.5 and 15.5 gestation days seem to allow a synergistic action of mutated p53 and c-Ha-Ras to overcome cell cycle arrest and facilitate the cell to pass through the whole cell cycle. Presumably, the epigenetic changes might comprise pathways involved in chromatin remodelling and/or the Ras/Raf/MEK/ERK pathway. Two of the candidates that are also important in embryonal development are the Wnt/catenin and the Hedgehog (HH) pathways. Since the HH pathway is also connected to the K-Ras pathway [18, 28] and plays a central role in the embryonal patterning during the phase under scrutiny, it is our prime candidate. HH regulates embryonal patterning through gradients of its 3 isoforms, however, in some adult tissues HH is also responsible for homeostasis and has effects on cell proliferation and apoptosis. Most importantly, deregulated HH can also lead to cancer development [1, 22, 33] and cyclopamine, an inhibitor of the HH pathway, is able to reduce metastasis [8, 9].
At 32˚C ts p53 adopts wt conformation and cells accumulate in G1 phase of the cell cycle. The ratio of cells in S phase was strongly reduced in all tested cells. The immortalized cells from young embryos (402/534) were nearly completely arrested in G1 phase after 24 h at 32˚C, whereas the immortalized cells from older embryos (602/534) showed a reduction in S phase, but not in G2 phase pointing to a different regulation in both cell types. However, transformed cells from oRECs showed a stronger response to the temperature shift. After shifting the cells back to 37˚C, transformed cells from oRECs re-entered the cell cycle much faster then transformed cells from yRECs. As expected, transformed cells entered the cell cycle more quickly than their immortalized counterparts.
The most salient finding of our present work is the strong impact of the endogenous cell traits in o vs y RECs. Our results show that even strong oncogenes such as mutated c-Ha-RAS and mutated TP53 are not able to override the intrinsic cellular program. Taken together, our results show that transformed RECs from older embryos show a higher growth potential than their counterparts from yRECs and are less susceptible to the action of CDK inhibitors. However, after inactivation of c-Ha-Ras with an inhibitor of farnesylation, also the transformed oRECs are strongly susceptible to growth inhibition by CDK inhibitors. If the phenotype of a certain tumor is known, this knowledge might help to develop a customized treatment for tumors with constitutively activated Ras.
Abbreviations
- CDKs:
-
cyclin-dependent kinases
- FPTase:
-
farnesylproteintransferase
- FTI:
-
inhibitors of FPTase
- HH:
-
Hedgehog
- MDM-2:
-
mouse double minute-2
- OLO:
-
olomoucine
- PARP-1:
-
poly(ADP-ribose) polymerase-1
- PD:
-
Petri dish
- PDT:
-
population doubling time
- PI:
-
propidium iodide
- REC:
-
rat embyonal cell
- ROSC:
-
roscovitine
- SD:
-
standard deviation
- ts:
-
temperature-sensitive
- wt:
-
wild-type
References
Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA (2003) Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 425(6960):846–851
Bernstein C, Bernstein H, Payne CM, Garewal H (2002) DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat. Res. 511(2):145–178
Blagosklonny MV (2002) P53: an ubiquitous target of anticancer drugs. Int. J. Cancer 98(2):161–166
De Laurenzi V, Costanzo A, Barcaroli D, Terrinoni A, Falco M, Annicchiarico-Petruzzelli M, Levrero M, Melino G (1998) Two new p73 splice variants, gamma and delta, with different transcriptional activity. J Exp Med 188(9):1763–1768
Dhavan R, Tsai LH (2001) A decade of CDK5. Nat. Rev. 2(10):749–759
Eliyahu D, Michalovitz D, Oren M (1985) Overproduction of p53 antigen makes established cells highly tumorigenic. Nature. 316(6024):158–160
Evans SC, Lozano G (1997) The Li-Fraumeni syndrome: an inherited susceptibility to cancer. Mol Med Today 3(9):390–395
Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, Karikari C, Alvarez H, Iacobuzio-Donahue C, Jimeno A, Gabrielson KL, Matsui W, Maitra A (2007) Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 67(5):2187–2196
Feldmann G, Habbe N, Dhara S, Bisht S, Alvarez H, Fendrich V, Beaty R, Mullendore M, Karikari C, Bardeesy N, Ouellette MM, Yu W, Maitra A (2008) Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 57(10):1420–1430
Gottlieb E, Haffner R, King A, Asher G, Gruss P, Lonai P, Oren M (1997) Transgenic mouse model for studying the transcriptional activity of the p53 protein: age- and tissue-dependent changes in radiation-induced activation during embryogenesis. Embo J 16(6):1381–1390
Greenblatt MS, Bennett WP, Hollstein M, Harris CC (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 54(18):4855–4878
Halevy O, Michalovitz D, Oren M (1990) Different tumor-derived p53 mutants exhibit distinct biological activities. Science. 250(4977):113–116
Harris CC (1996) p53 tumor suppressor gene: at the crossroads of molecular carcinogenesis, molecular epidemiology, and cancer risk assessment. Environ. Health Perspect. 104(Suppl 3):435–439
Havlicek L, Hanus J, Vesely J, Leclerc S, Meijer L, Shaw G, Strnad M (1997) Cytokinin-derived cyclin-dependent kinase inhibitors: synthesis and cdc2 inhibitory activity of olomoucine and related compounds. J. of Med. Chem. 40(4):408–412
Ide F, Kitada M, Sakashita H, Kusama K, Tanaka K, Ishikawa T (2003) p53 haploinsufficiency profoundly accelerates the onset of tongue tumors in mice lacking the xeroderma pigmentosum group A gene. Am. J. Pathol. 163(5):1729–1733
Inga A, Storici F, Darden TA, Resnick MA (2002) Differential transactivation by the p53 transcription factor is highly dependent on p53 level and promoter target sequence. Mol. Cell. Biol. 22(24):8612–8625
Janicke RU, Sohn D, Schulze-Osthoff K (2008) The dark side of a tumor suppressor: anti-apoptotic p53. Cell Death Differ. 15(6):959–976
Ji Z, Mei FC, Xie J, Cheng X (2007) Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem 282(19):14048–14055
Kanai M, Hanashiro K, Kim SH, Hanai S, Boulares AH, Miwa M, Fukasawa K (2007) Inhibition of Crm1–p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat. Cell Biol. 9(10):1175–1183
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW (1991) Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 51(23 Pt 1):6304–6311
Kolch W (2000) Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 351(Pt 2):289–305
Lau J, Kawahira H, Hebrok M (2006) Hedgehog signaling in pancreas development and disease. Cell. Mol. Life Sci. 63(6):642–652
Li FP, Fraumeni JF Jr (1969) Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Intern. Med. 71(4):747–752
Liu L, Guo J, Yuan L, Cheng M, Cao L, Shi H, Tong H, Wang N, De W (2007) Alpha-fetoprotein is dynamically expressed in rat pancreas during development. Dev. Growth Differ. 49(8):669–681
Michalovitz D, Halevy O, Oren M (1990) Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell 62(4):671–680
Offer H, Wolkowicz R, Matas D, Blumenstein S, Livneh Z, Rotter V (1999) Direct involvement of p53 in the base excision repair pathway of the DNA repair machinery. FEBS Lett. 450(3):197–204
Paglini G, Caceres A (2001) The role of the Cdk5–p35 kinase in neuronal development. European journal of biochemistry / FEBS 268(6):1528–1533
Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M (2006) Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 20(22):3161–3173
Schmid G, Kramer MP, Maurer M, Wandl S, Wesierska-Gadek J (2007) Cellular and organismal ageing: Role of the p53 tumor suppressor protein in the induction of transient and terminal senescence. J. Cell. Biochem. 101(6):1355–1369
Schmid G, Kramer MP, Wesierska-Gadek J (2009) p53-mediated regulation of cell cycle progression: pronounced impact of cellular microenvironment. J. Cell. Physiol. 219(2):459–469
Taurin S, Seyrantepe V, Orlov SN, Tremblay TL, Thibault P, Bennett MR, Hamet P, Pshezhetsky AV (2002) Proteome analysis and functional expression identify mortalin as an antiapoptotic gene induced by elevation of [Na+]i/[K+]i ratio in cultured vascular smooth muscle cells. Circ. Res. 91(10):915–922
Taylor WR, Egan SE, Mowat M, Greenberg AH, Wright JA (1992) Evidence for synergistic interactions between ras, myc and a mutant form of p53 in cellular transformation and tumor dissemination. Oncogene 7(7):1383–1390
Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M (2003) Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 425(6960):851–856
Toledo F, Wahl GM (2006) Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 6(12):909–923
Viktorsson K, De Petris L, Lewensohn R (2005) The role of p53 in treatment responses of lung cancer. Biochem. Biophys. Res. Commun. 331(3):868–880
Vindelov LL, Christensen IJ, Nissen NI (1983) A detergent-trypsin method for the preparation of nuclei for flow cytometric DNA analysis. Cytometry 3(5):323–327
Wadhwa R, Takano S, Kaur K, Deocaris CC, Pereira-Smith OM, Reddel RR, Kaul SC (2006) Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 118(12):2973–2980
Wesierska-Gadek J, Schmid G (2000) Overexpressed poly(ADP-ribose) polymerase delays the release of rat cells from p53-mediated G(1) checkpoint. J. Cell. Biochem. 80(1):85–103
Wesierska-Gadek J, Schmid G (2001) Poly(ADP-ribose) polymerase-1 regulates the stability of the wild-type p53 protein. Cell. Mol. Biol. Lett. 6(2):117–140
Wesierska-Gadek J, Bugajska-Schretter A, Cerni C (1996a) ADP-ribosylation of p53 tumor suppressor protein: mutant but not wild- type p53 is modified. J Cell Biochem 62(1):90–101
Wesierska-Gadek J, Schmid G, Cerni C (1996b) ADP-ribosylation of wild-type p53 in vitro: binding of p53 protein to specific p53 consensus sequence prevents its modification. Biochemical and biophysical research communications 224(1):96–102
Wesierska-Gadek J, Wojciechowski J, Schmid G (2003) Central and carboxy-terminal regions of human p53 protein are essential for interaction and complex formation with PARP-1. J. Cell. Biochem. 89(2):220–232
Wesierska-Gadek J, Wojciechowski J, Schmid G (2003) Phosphorylation regulates the interaction and complex formation between wt p53 protein and PARP-1. J. Cell. Biochem. 89(6):1260–1284
Wesierska-Gadek J, Gueorguieva M, Ranftler C, Zerza-Schnitzhofer G (2005) A new multiplex assay allowing simultaneous detection of the inhibition of cell proliferation and induction of cell death. J. Cell. Biochem. 96(1):1–7
Wesierska-Gadek J, Kramer M, Schmid G (2006) Prevention of farnesylation of c-Ha-Ras protein enhances synergistically the cytotoxic action of doxorubicin in cycling but not in quiescent cells. J. Cell. Biochem. 99(6):1664–1676
Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F (1998) p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 2(3):305–316
Zhang Z, Yao R, Li J, Wang Y, Boone CW, Lubet RA, You M (2005) Induction of invasive mouse skin carcinomas in transgenic mice with mutations in both H-ras and p53. Mol Cancer Res 3(10):563–574
Zhang Z, Wang Y, Yao R, Li J, Lubet RA, You M (2006) p53 Transgenic mice are highly susceptible to 4-nitroquinoline-1-oxide-induced oral cancer. Mol Cancer Res 4(6):401–410
Acknowledgements
The paper was partially supported by a grant from the Austrian Funding Agency FWF (P19894-B11).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Węsierska-Gądek, J., Walzi, E., Dolečkova, I. et al. Oncogenes do not Fully Override Cell-intrinsic Traits: Pronounced Impact of the Cellular Programme. Cancer Microenvironment 2 (Suppl 1), 215–225 (2009). https://doi.org/10.1007/s12307-009-0024-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-009-0024-9