
Abstract
Melanoma growth, angiogenesis and metastatic progression are strongly promoted by the inflammatory tumor microenvironment due to high levels of cytokine and chemokine secretion by the recruited inflammatory and stromal cells. In addition, platelets and molecular components of procoagulant pathways have been recently emerging as critical players of tumor growth and metastasis. In particular, thrombin, through the activity of its receptor protease-activated receptor-1 (PAR-1), regulates tumor cell adhesion to platelets and endothelial cells, stimulates tumor angiogenesis, and promotes tumor growth and metastasis. Notably, in many tumor types including melanoma, PAR-1 expression directly correlates with their metastatic phenotype and is directly responsible for the expression of interleukin-8, matrix metalloproteinase-2 (MMP-2), vascular endothelial growth factor, platelet-derived growth factor, and integrins. Another proinflammatory receptor–ligand pair, platelet-activating factor (PAF) and its receptor (PAFR), have been shown to act as important modulators of tumor cell adhesion to endothelial cells, angiogenesis, tumor growth and metastasis. PAF is a bioactive lipid produced by a variety of cells from membrane glycerophospholipids in the same reaction that releases arachidonic acid, and can be secreted by platelets, inflammatory cells, keratinocytes and endothelial cells. We have demonstrated that in metastatic melanoma cells, PAF stimulates the phosphorylation of cyclic adenosine monophosphate response element-binding protein (CREB) and activating transcription factor 1 (ATF-1), which results in overexpression of MMP-2 and membrane type 1-MMP (membrane type 1-MMP). Since only metastatic melanoma cells overexpress CREB/ATF-1, we propose that metastatic melanoma cells are better equipped than their non-metastatic counterparts to respond to PAF within the tumor microenvironment. The evidence supporting the hypothesis that the two G-protein coupled receptors, PAR-1 and PAFR, contribute to the acquisition of the metastatic phenotype of melanoma is presented and discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The development of tumor metastasis is a complex cascade of events [1]. Potentially, metastatic cells have to exit the primary tumor site by loosening cell to cell contact, adhering to and degrading extracellular matrix (ECM), migrating through the subendothelial basement membrane of local post-capillary veins and lymphatic vessels and intravasating. Once in circulation, tumor cells face severe mechanical and immunosurveillance challenges. Surviving cells can arrest in the peripheral capillary bed of a distant organ, adhere to the subendothelial basement membrane, extravasate, adhere and migrate through the ECM, and form a colony at the new metastatic site. Further induction of neoangiogenesis must occur to assure continuous growth [1].
An expending amount of data reveals the importance of an inflammatory microenvironment and stroma in cancer initiation and progression, which brings new directions and approaches to cancer treatment [2–8]. Genetic and functional experiments indicated that inflammatory cells such as tumor-infiltrating monocytes/macrophages, neutrophils, mast cells, eosinophils and activated T and B lymphocytes, as well as stromal fibroblasts contribute to malignancies by releasing growth and survival factors, as well as extracellular proteases, and other proangiogenic factors [2–7]. Melanoma, as with all other cancers, is comprised of a group of heterogeneous cells that co-exist and interact with an infrastructure of other cells (keratinocytes, fibroblasts, endothelial cells, inflammatory cells) and extracellular matrix components (laminin, collagen), growth factors (vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), thrombin), as well as proteases and interleukins involved in invasion (matrix metalloproteinases (MMPs), interleukin-8 (IL-8), urokinase-type plasminogen activator) [9–11]. As described by Haass and Herlyn [9], alterations in keratinocyte-mediated contact growth inhibition in the skin may drive the malignant transformation of melanocytes. Keratinocytes that are activated by ultraviolet (UV) for example, release interleukins 1, 3, 6 and 8, secrete tumor necrosis factor-α (TNF-α), granulocyte-macrophage colony-stimulating factor, and generate the proinflammatory biolipid platelet-activating factor (PAF), contributing to melanoma development and progression [12–16]. Within the tumor microenvironment, a rapid proliferation of fibroblasts, whose role has been neglected previously, is supported by platelet-derived growth factor (PDGF), bFGF and transforming growth factor-β (TGF-β) produced by melanoma as well as inflammatory cells [10, 17]. In turn, fibroblasts produce a series of growth factors such as insulin-like growth factor-1, hepatocyte growth factor (HGF)/scatter factor, bFGF, and TGF-β that further support the growth and proliferation of melanoma cells [10, 17]. Recently, fibroblasts have been shown to remodel the matrix and form “tracks” creating a leading edge for tumor cells invasion [18]. Strong association has been shown between the recruitment of Tie-2 (an angiopoietin receptor)-expressing monocytes and tumor angiogenesis, and between MMP-9-secreting tumor associated macrophages and tumor cell invasion [18–21]. In addition, platelets and molecular components of the coagulation system and platelet activation pathways have been emerging as critical players of tumor growth and metastasis [22–24]. Overall, it is now believed that cancer cells initiate a pathological cycle, whereby through the constitutive activity of major inflammatory signaling pathways such as nuclear factor-κB (NF-κB), signal transducer and activator of transcription 3 (STAT3) and cyclooxygenases-2 (COX-2)-driven lipid metabolism, they trigger the recruitment and activation of the inflammatory cells, stroma and platelets [3–5, 25–29]. In turn, cells within the tumor microenvironment produce inflammatory mediators and angiogenic factors or adhesion-stimulating factors to further amplify the metastatic phenotype [3–5, 25–29].
The progression of melanoma from radial growth phase to vertical growth phase and metastatic dissemination are accompanied by multiple molecular changes [30]. This review will focus on the role of molecular intermediates of the coagulation and platelet activation pathways in tumor metastasis. Specifically, we will focus on the thrombin receptor protease-activated receptor (PAR-1) and the platelet-activating factor receptor (PAFR). We will provide evidence for sensitization of metastatic melanoma cells to the stimulatory effects of thrombin and PAF.
Role of Coagulation and Platelet Activation in Tumor Growth and Metastasis
Tumor cells produce and activate the components of the coagulation/platelet activation pathways—thrombin, tissue factor, fibrinogen, von Willebrand factor (VWF) and PAF [23, 31, 32]. Thrombin is an essential component of the tumor microenvironment already present at the primary tumor site because of the leaky nature of the tumor vasculature. There, the migration of tumor cells into the vasculature is stimulated by thrombin, which is induced by tissue factor on the surface of most tumor cells [23].
Tumor–host cell interaction becomes vital for the survival of potentially metastatic cells during vascular dissemination. In the circulation, thrombin and other tumor-secreted agents activate endothelial cells to express P-selectin on their surface. P-selectin binds weakly to tumor cells containing the P-selectin ligand receptor. Weakly activated platelets also bind tumor cells via P-selectins. This induces weak tethering of tumor to the endothelium and platelets [23, 31–34]. A tighter combination of platelets and tumor cells develops, which produces thrombin at a more rapid rate, since platelets provide a catalytic surface for thrombin generation from coagulant proteins. This leads to a firm bond between platelets and tumor cells, mediated by platelet integrin IIb–IIIa binding to tumor integrins via VWF, fibronectin, and other RGDS-domain containing ligands [23]. Platelet activation also leads to angiogenesis via thrombin-stimulated synthesis and secretion of VEGF and growth related oncogene-α from tumor cells, as well as release of PDGF, VEGF, and angiopoietin-1 from platelets, and increased angiopoietin-2 and kinase insert-domain-containing receptor in endothelial cells [35–37]. In addition, stimulated platelets are an important source of growth and angiogenesis-inducing biolipids lysophosphatidic acid and PAF [38]. Besides mediating adhesion to endothelial cells and development of collateral vessels, platelet-tumor aggregates protect tumor cells from natural killer cells [39]. These aggregates further embolize, leading to ischemia and endothelial cell denudation [23]. As a result, tumor cells and platelets bind more avidly to the subendothelial basement membrane and matrix. Finally, tumor emboli lead to tumor extravasation into the parenchyma and neoangiogenesis [23].
Thrombin is also a prominent angiogenic factor. It promotes endothelial cell alignment in Matrigel in vitro and angiogenesis in vivo [40]. It induces the differentiation of endothelial cells into capillary structures on Matrigel and increases endothelial cell migration [40].
The Role of PAR-1 in Tumor Growth and Metastasis
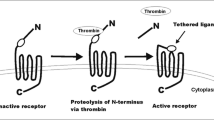
Thrombin not only stimulates platelets and induces angiogenesis, it also directly activates tumor cells through the activity of its receptor PAR-1. The thrombin receptor is a seven transmembrane-spanning G-protein coupled receptor. Unlike typical ligand receptor interactions, thrombin does not activate PAR-1 upon binding. Rather, it cleaves the N-terminus of PAR-1 at serine 42. Upon cleavage, the new amino terminal peptide acts as a tethered ligand that will now bind to the body of the receptor thereby causing cell signaling via G-proteins. As mentioned above, in order to activate thrombin, melanoma and other tumor cells constitutively express tissue factor (TF) [31, 32]. The hypoxic tumor microenvironment also induces TF expression by endothelial cells, tumor associated macrophages and myofibroblasts thereby also augmenting thrombin production in the tumor microenvironment [41]. PAR-1 can also be activated by ligands other than thrombin such as factor Xa, granzyme A, trypsin and plasmin [33, 42, 43]. It has also been reported recently that PAR-1 in breast cancer cells can be proteolytically cleaved and activated by MMP-1 [44].
In tumor cells, PAR-1 stimulates expression of adhesion molecules such as integrins αIIbβ3, αvβ5, and αvβ3 [45–47]. Indeed, thrombin-treated melanoma cells enhance their adhesion to platelets and fibronectin in vitro [48]. In various types of cells, including vascular endothelial cells, PAR-1 activation results in upregulation of gene products involved in invasion (MMP-2) [49], and angiogenesis (IL-8, VEGF, bFGF, PDGF) [50–53]. In human melanoma cells, thrombin acts as a growth factor and is mitogenic [32]. Overall, thrombin and PAR-1 contribute to the acquisition of the metastatic phenotype of melanoma by facilitating tumor invasion and metastasis through the induction of cell adhesion molecules, matrix degrading proteases, and stimulating the secretion of angiogenic factors into the melanoma tumor microenvironment (Fig. 1).

Schematic representation of molecules involved in cell invasion and angiogenesis via activation of PAR-1 which is overexpressed in metastatic melanoma cells. Thrombin from the microenvironment cleaves the N-terminus of PAR-1 to activate the receptor. The tumor-promoting signals transduced by PAR-1 through G-proteins upregulate molecules involved in angiogenesis and invasion
Our tissue analysis from patients demonstrated that PAR-1 is overexpressed predominantly in malignant melanoma tumors and in metastatic lesions as compared to common melanocytic nevi and normal skin [54]. Furthermore, a significantly higher percentage of PAR-1 positive cells was found in metastatic melanoma specimens as compared to both dysplastic nevi and primary melanoma specimens [55]. In addition to melanoma, overexpression of PAR-1 has been observed in a variety of human cancers such as breast, lung, colon, pancreatic and prostate [44, 56–59]. We further demonstrated that PAR-1 is overexpressed in highly metastatic melanoma cell lines as compared to non-metastatic ones [34, 60]. We found that the overexpression of PAR-1 in the highly invasive and aggressive melanoma cell lines correlates with the loss of the activator protein-2α (AP-2α) transcription factor, which is a crucial event in the progression of human melanoma [60]. An inverse correlation between AP-2α and PAR-1 expression was also established using a “progressive” melanoma tissue microarray [55].
The Role of PAFR in Melanoma Progression
Activated platelets release another proangiogenic biolipid-platelet-activating factor. PAF is a potent proinflammatory mediator and the first bioactive lipid ever identified [61–64]. PAF may be produced from the 1-O-alkyl-2-arachidonoyl-sn-glycero-3-phosphocholine via enzymatic hydrolysis catalyzed by phospholipase A2 [65–69]. In a parallel process, the arachidonic acid released in the first reaction is further converted by the COX-2 enzyme to form the precursor of prostanoids. PAF acts through the specific seven transmembrane-spanning G-protein-coupled receptor PAFR [70–72].
Studies on inflammatory cells, endothelial cells, keratinocytes, and various other types of cells demonstrated that a long lasting consequence of transient stimulation with highly unstable PAF and PAF-like biolipids is the expression of inflammatory cytokines and mediators such as IL-6, IL-8, IL-10, COX-2, VEGF and inducible nitric oxide synthase (Table 1) [24–26]. An essential role of PAF as a second messenger in endothelial and inflammatory cells was further established following the findings that lipopolysaccharide-, IL-1- and TNF-α- induced activation of NF-κB is a PAF-dependent process [27].
PAF and Angiogenesis
PAF is a potent mediator of tumor neoangiogenesis [73–79]. Numerous reports showed that PAF can activate endothelial cells directly, as well as mediate angiogenesis induced by other angiogenic factors [74, 80]. Camussi’s group demonstrated that PAF produced by breast or Kaposi’s sarcoma cancer cells induces and sustains the in vivo neoangiogenesis in experimental tumor models [80, 81]. Robert and Hunt [79] has demonstrated the effectiveness of PAF antagonists in the inhibition of angiogenesis in prostate cancer xenografts. The same group showed that PAF induces activation of matrix metalloproteinase-2 activity and vascular endothelial cell invasion and migration [82]. They further found that bFGF stimulated PAF-dependent proliferation in human umbilical vein endothelial cells (HUVEC) [83]. HGF and TNF-α both induce angiogenesis through mechanisms that involve the production of PAF [75, 77, 78]. Montrucchio et al. [77] demonstrated that nitric oxide mediates the angiogenesis induced by PAF or TNF, the latter itself being dependent on the production of PAF. Brizzi et al. [73] showed that HUVECs express the thrombopoietin receptor, which activates cell migration in vitro and angiogenesis in vivo; these effects were mediated by PAF- and IL-8-dependent phosphorylation of STAT1 and STAT5B. Furthermore, it was found that vascular permeability induced by VEGF was mediated by PAF [84]. In HUVECs, both VEGF-induced P-selectin translocation and subsequent neutrophil adhesion requires PAF synthesis [85]. Moreover, angiopoietin-1, and -2 stimulate PAF synthesis [86].
PAF and PAF Receptor in Melanoma
The first demonstration of the role of PAF in melanoma metastasis was made in 1996 by Im et al. [87]. They found that IL-1α and TNF-α-induced increase in experimental pulmonary metastasis produced by the B16F10 murine melanoma cells in C57Bl/6 mice was augmented by a single intraperitoneal injection of PAF [87]. Several repeated injections of PAFR antagonist BN50739 (day 0 through 2) decreased both IL-1α and TNF-α-induced metastasis as well as control lung metastasis, suggesting the role of endogenous PAF in tumor cell lung colonization. PAF caused an increase in the retention of radiolabeled B16F10 cells in the lungs, suggesting that stimulation of endothelial cell adhesion was the primary mechanism for the observed pro-metastatic effect of PAF [87].
PAFR transgenic mice exhibited progressive hyperproliferative changes in the epidermis as soon as 2 weeks after birth. The keratinocyte hyperplasia was accompanied by hyperpigmentation and an increase in the number of dermal melanocytes in the ear and tail, with consequent development of melanocytic tumors late in life [88, 89]. The PAFR transgene expression was detected in keratinocytes but not in melanocytes, suggesting that the progressive recruitment of melanocytes to the dermis was driven by keratinocytes, and possibly by the accumulating fibroblasts and mast cells. In human skin, all these types of cells are indeed known to play a significant role in regulating skin homeostasis and behavior of resident melanocytes, as well as melanoma growth and local malignant invasion [10].
In human skin, the melanocyte homeostasis and number is tightly controlled by neighboring keratinocytes through an E-cadherin-mediated adhesion [10]. Essential for melanoma tumorigenesis, keratinocytes and corneal stromal cells secrete PAF in response to UV exposure [13, 14, 90–92]. Keratinocytes express PAF receptors on their surface [93], and PAF upregulates their COX-2, IL-6, and IL-8 mRNA expression and prostaglandin E2 secretion [14, 15]. Although the incidence of severe sunburns in childhood have been linked to melanoma development later in life, the precise mechanism by which UV contributes to melanomagenesis has not been discovered. This mechanism may involve, at least in part, UV-induced immunosuppression [94, 95]. Interestingly, Walterscheid et al. [15] have found that the UVB-induced inflammation in mouse skin and the consequent systemic immunosuppression was abolished by PAFR antagonists. Indeed, Travers’s group has demonstrated UVB-induced generation of PAF-like substances in human epidermal keratinocytes [13]. Furthermore, staphylococcal lipoteichoic acid was also found to inhibit the delayed-type hypersensitivity reactions via direct binding to the platelet-activating factor receptor [96]. It is therefore possible that PAF and other PAFR agonists are not only important for melanoma cell biology, but that they may play a pivotal role in tumor growth by ‘negatively’ modulating the immune system and inhibiting Th1 cytotoxic responses.
Further support for the role of PAF receptor in melanoma growth and metastatic dissemination came from the experimental systems utilizing in vitro and in vivo murine melanoma models. Indeed, it was reported that inhibition of PAF activity by means of overexpressing PAF-acetyl-hydrolase in B16F10 murine melanoma cells led to a significant decrease in tumor vascularization and growth, allowing longer animal survival [97]. Most recently, Fallani et al. have demonstrated that PAF is being synthesized by B16F10 cells in response to interferon-γ treatment, and that PAF promoted the invasion of these cells through the Matrigel-coated filters [98]. Moreover, it has been shown that a single intraperitoneal injection of PAF induced expression of MMP-9 and MMP-2 in the mouse lungs, and significantly enhanced B16F10 pulmonary lung metastasis, suggesting an additional, non-tumor-cell mechanism of PAF action [99]. Authors further showed that selective inhibition of MMP-9, which was expressed by bronchial epithelial cells as well as in the walls of blood vessels after stimulation with PAF, completely prevented B16F10 metastasis, whereas selective inhibition of MMP-2 was insufficient [99]. Notably, MMP-9 is expressed predominantly by tumor stroma, where it is believed to play a critical role in tumor invasion and extracellular growth factor activation. Indeed, in mouse models of squamous carcinogenesis, MMP- 9 was predominantly found in neutrophils, macrophages, and mast cells, rather than in oncogene-positive neoplastic cells [21]. In the human melanoma cell line Hs294T, PAFR antagonists were able to prevent adhesion to IL-1-stimulated endothelial cells [100].
Our in vivo experiments showed that the PAF receptor antagonist PCA4248 acts as a potent inhibitor of experimental melanoma human lung metastasis when delivered intravenously before melanoma cell inoculation. Furthermore, daily treatments with PCA4248 inhibited growth of established microscopic tumor cell colonies in the lungs. This indicates that antagonizing PAFR activity could serve as a point of intervention during vascular dissemination or tumor/metastasis outgrowth [101].
PAF Induces MMP-2 and MT1-MMP in Melanoma in part via Activation of CREB/ATF-1
Recently, we have examined the role of PAF in human melanoma metastasis and found that PAF receptor was expressed in all cultured melanoma cell lines regardless of their metastatic potential [101]. In metastatic melanoma cell lines, which are known to overexpress cyclic adenosine monophosphate (cAMP)-response element-binding protein (CREB) and activating transcription factor-1 (ATF-1) transcription factors, PAF induced CREB and ATF-1 phosphorylation as well as secretion and activation of MMP-2 and membrane type 1 (MT1)-MMP via a PAFR-mediated signal transduction mechanism that required pertussis toxin-insensitive Gαq protein and adenylate cyclase activity and was antagonized by a cAMP-dependent protein kinase A (PKA) and p38 mitogen activated protein kinase (MAPK) inhibitors [101]. Other kinases and transcription factors may be also involved in PAF-induced activation of MMP-2, such as janus kinase-2-Src-STAT-3 regulatory node, as demonstrated in HUVEC cells [83, 102]. We propose that all melanoma cells, regardless of their metastatic potential, express PAFR and secrete basal levels of MMP-2 and MT1-MMP. However, within the melanoma tumor microenvironment, where melanoma cells come into contact with PAF-secreting cells such as platelets, endothelial cells, and inflammatory cells, PAF will phosphorylate CREB and ATF-1 through the activity of its receptor and a signaling cascade involving p38 MAPK and PKA. This mechanism, as well as other possible regulatory pathways will further result in overexpression and secretion of MMP-2 and MT1-MMP (Fig. 2). However, since only metastatic melanoma cells overexpress CREB and ATF-1, they are, therefore, better equipped to respond to the effect of PAF within the tumor microenvironment.

A model for the stimulation of MMP-2 and MT1-MMP by PAF via activation of CREB/ATF-1. We propose that melanoma cells, regardless of their metastatic potential, express PAFR and secrete basal levels of MMP-2 and MT1-MMP. However, within the melanoma tumor microenvironment, melanoma cells come into contact with platelets, endothelial cells, and inflammatory cells that secrete PAF. PAF, through the activity of its receptor on tumor cells and a signaling cascade involving pertussis-toxin-insensitive Gαq protein, adenylate cyclase, p38 MAPK and PKA, phosphorylates CREB and ATF-1. Activation of this and possibly other signaling mechanisms results in overexpression and secretion of MMP-2 and MT1-MMP. However, since only metastatic melanoma cells overexpress CREB and ATF-1, they are better equipped to respond to the stimulatory effect of PAF within the tumor microenvironment
Conclusion
It is apparent that interactions between tumor cells and the components of the coagulation and platelet activation pathways is critical for tumor growth and metastatic dissemination. Through the activity of their specific receptors on various types of cells including platelets, endothelial and tumor cells, thrombin and platelet-activating factor promote inflammatory and angiogenic responses, the remodeling of the extracellular matrix as well as the process of vascular dissemination. PAR-1 is overexpressed in metastatic melanoma cells. Its activation by thrombin promotes secretion of adhesion, angiogenic and survival factors into the tumor microenvironment allowing for increased metastatic potential of melanoma. Simultaneously, many inflammatory stimuli can trigger production of PAF by an array of cells, including tumor cells. Activated PAFR causes stimulation of the CREB and ATF-1 transcription factors in melanoma, which in turn increase the secretion of MMP-2 and MT1-MMP. Since metastatic melanoma cells overexpress PAR-1 as well as CREB/ATF-1 downstream of PAFR signaling pathway, we speculate that metastatic melanoma cells are better equipped to respond to inflammatory stimulation from the tumor microenvironment.
Abbreviations
- PAR-1:
-
protease-activated receptor-1
- IL-8:
-
interleukin-8
- MMP-2:
-
matrix metalloproteinase-2
- VEGF:
-
vascular endothelial growth factor
- PDGF:
-
platelet-derived growth factor
- PAF:
-
platelet-activating factor
- PAFR:
-
PAF receptor
- CREB:
-
cyclic AMP-response element-binding protein
- ATF-1:
-
activating transcription factor-1
- ECM:
-
extracellular matrix
- bFGF:
-
basic fibroblast growth factor
- uPA:
-
urokinase-type plasminogen activator
- TNF-α:
-
tumor necrosis factor-α
- GM-CSF:
-
granulocyte-macrophage colony-stimulating factor
- TGF-β:
-
transforming growth factor-β
- IGF-1:
-
insulin-like growth factor-1
- HGS/SF:
-
hepatocyte growth factor/scatter factor
- GRO-α:
-
growth related oncogene-α
- NK cells:
-
natural killer cells
- TF:
-
tissue factor
- AP-2α:
-
activator protein-2α
- STAT:
-
signal transducer and activator of transcription
- PKA:
-
protein kinase A
References
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3(6):453–458
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow. Lancet 357(9255):539–545
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420(6917):860–867
Mantovani A (2005) Cancer: inflammation by remote control. Nature 435(7043):752–753
Pikarsky E et al (2004) NF-kappa B functions as a tumour promoter in inflammation-associated cancer. Nature 431(7007):461–466
Smalley KS, Lioni M, Herlyn M (2005) Targeting the stromal fibroblasts: a novel approach to melanoma therapy. Expert Rev Anticancer Ther 5(6):1069–1078
Denardo DG, Johansson M, Coussens LM (2007) Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev DOI 10.1007/s10555-007-9100-0
Tlsty TD, Coussens LM (2006) Tumor stroma and regulation of cancer development. Annu Rev Pathol 1:119–150
Haass NK, Herlyn M (2005) Normal human melanocyte homeostasis as a paradigm for understanding melanoma. J Investig Dermatol Symp Proc 10(2):153–163
Lee JT, Herlyn M (2006) Microenvironmental influences in melanoma progression. J Cell Biochem 101(4):862–872
Postovit LM et al (2006) Influence of the microenvironment on melanoma cell fate determination and phenotype. Cancer Res 66(16):7833–7836
Schwarz T, Luger TA (1989) Effect of UV irradiation on epidermal cell cytokine production. J Photochem Photobiol B 4(1):1–13
Marathe GK et al (2005) Ultraviolet B radiation generates platelet-activating factor-like phospholipids underlying cutaneous damage. J Biol Chem 280(42):35448–35457
Pei Y et al (1998) Activation of the epidermal platelet-activating factor receptor results in cytokine and cyclooxygenase-2 biosynthesis. J Immunol 161(4):1954–1961
Walterscheid JP, llrich SE, Nghiem DX (2002) Platelet-activating factor, a molecular sensor for cellular damage, activates systemic immune suppression. J Exp Med 195(2):171–179
Ullrich SE (2005) Mechanisms underlying UV-induced immune suppression. Mutat Res 571(1–2):185–205
Hsu MY, Meier F, Herlyn M (2002) Melanoma development and progression: a conspiracy between tumor and host. Differentiation 70(9–10):522–536
Gaggioli C et al (2007) Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9(12):1392–1400
De Palma M et al (2005) Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8(3):211–226
Lewis CE, De Palma M, Naldini L (2007) Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res 67(18):8429–8432
Coussens LM et al (2000) MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103(3):481–490
Gasic GJ, Gasic TB, Stewart CC (1968) Antimetastatic effects associated with platelet reduction. Proc Natl Acad Sci U S A 61(1):46–52
Nierodzik ML, Karpatkin S (2006) Thrombin induces tumor growth, metastasis, and angiogenesis: evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 10(5):355–362
Nierodzik ML et al (1998) Protease-activated receptor 1 (PAR-1) is required and rate-limiting for thrombin-enhanced experimental pulmonary metastasis. Blood 92(10):3694–3700
Gupta RA, Dubois RN (2001) Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 1(1):11–21
Affara NI, Coussens LM (2007) IKK alpha at the crossroads of inflammation and metastasis. Cell 129(1):25–26
Brown JR, DuBois RN (2004) Cyclooxygenase as a target in lung cancer. Clin Cancer Res 10(12 Pt 2):4266s–4269s
Dauer DJ et al (2005) Stat3 regulates genes common to both wound healing and cancer. Oncogene 24(21):3397–3408
Robinson SC, Coussens LM (2005) Soluble mediators of inflammation during tumor development. Adv Cancer Res 93:159–187
Nyormoi O, Bar-Eli M (2003) Transcriptional regulation of metastasis-related genes in human melanoma. Clin Exp Metastasis 20(3):251–263
Bromberg ME et al (1995) Tissue factor promotes melanoma metastasis by a pathway independent of blood coagulation. Proc Natl Acad Sci U S A 92(18):8205–8209
Fischer EG, Ruf W, Mueller BM (1995) Tissue factor-initiated thrombin generation activates the signaling thrombin receptor on malignant melanoma cells. Cancer Res 55(8):1629–1632
Ruf W, Mueller BM (2006) Thrombin generation and the pathogenesis of cancer. Semin Thromb Hemost 32(Suppl 1):61–68
Tellez C, Bar-Eli M (2003) Role and regulation of the thrombin receptor (PAR-1) in human melanoma. Oncogene 22(20):3130–3137
Kepner N, Lipton A (1981) A mitogenic factor for transformed fibroblasts from human platelets. Cancer Res 41(2):430–432
Mohle R et al (1997) Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci U S A 94(2):663–668
Li JJ et al (2001) Thrombin induces the release of angiopoietin-1 from platelets. Thromb Haemost 85(2):204–206
Boucharaba A et al (2004) Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest 114(12):1714–1725
Nieswandt B et al (1999) Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 59(6):1295–1300
Haralabopoulos GC et al (1997) Thrombin promotes endothelial cell alignment in Matrigel in vitro and angiogenesis in vivo. Am J Physiol 273(1 Pt 1):C239–C245
Ruf W (2007) Tissue factor and PAR signaling in tumor progression. Thromb Res 120(Suppl 2):S7–S12
Hansen KK, Saifeddine M, Hollenberg MD (2004) Tethered ligand-derived peptides of proteinase-activated receptor 3 (PAR3) activate PAR1 and PAR2 in Jurkat T cells. Immunology 112(2):183–190
O’Brien PJ et al (2001) Protease activated receptors: theme and variations. Oncogene 20(13):1570–1581
Boire A et al (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120(3):303–313
Even-Ram SC et al (2001) Tumor cell invasion is promoted by activation of protease activated receptor-1 in cooperation with the alpha v beta 5 integrin. J Biol Chem 276(14):10952–10962
Wojtukiewicz MZ et al (1992) Thrombin enhances tumor cell adhesive and metastatic properties via increased alpha IIb beta 3 expression on the cell surface. Thromb Res 68(3):233–245
Senger DR et al (1996) Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the alphavbeta3 integrin, osteopontin, and thrombin. Am J Pathol 149(1):293–305
Nierodzik ML et al (1996) Presence of the seven transmembrane thrombin receptor on human tumour cells: effect of activation on tumour adhesion to platelets and tumor tyrosine phosphorylation. Br J Haematol 92(2):452–457
Zucker S et al (1995) Thrombin induces the activation of progelatinase A in vascular endothelial cells. Physiologic regulation of angiogenesis. J Biol Chem 270(40):23730–23738
Shimizu S et al (2000) Thrombin stimulates the expression of PDGF in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 279(3):L503–L510
Ueno A et al (1996) Thrombin stimulates production of interleukin-8 in human umbilical vein endothelial cells. Immunology 88(1):76–81
Cucina A et al (1999) Thrombin induces production of growth factors from aortic smooth muscle cells. J Surg Res 82(1):61–66
Huang YQ et al (2001) Thrombin induces increased expression and secretion of VEGF from human FS4 fibroblasts, DU145 prostate cells and CHRF megakaryocytes. Thromb Haemost 86(4):1094–1098
Massi D et al (2005) Expression of protease-activated receptors 1 and 2 in melanocytic nevi and malignant melanoma. Hum Pathol 36(6):676–685
Tellez CS et al (2007) Quantitative analysis of melanocytic tissue array reveals inverse correlation between activator protein-2alpha and protease-activated receptor-1 expression during melanoma progression. J Invest Dermatol 127(2):387–393
Even-Ram S et al (1998) Thrombin receptor overexpression in malignant and physiological invasion processes. Nat Med 4(8):909–914
Kaushal V et al (2006) Thrombin receptor expression is upregulated in prostate cancer. Prostate 66(3):273–282
Rudroff C et al (1998) Characterization of functional thrombin receptors in human pancreatic tumor cells (MIA PACA-2). Pancreas 16(2):189–194
Wojtukiewicz MZ et al (1995) Solid tumor cells express functional “tethered ligand” thrombin receptor. Cancer Res 55(3):698–704
Tellez C et al (2003) Loss of activator protein-2alpha results in overexpression of protease-activated receptor-1 and correlates with the malignant phenotype of human melanoma. J Biol Chem 278(47):46632–46642
Benveniste J et al (1979) Semi-synthesis and proposed structure of platelet-activating factor (P.A.F.): PAF-acether an alkyl ether analog of lysophosphatidylcholine. C R Seances Acad Sci D 289(14):1037–1040
Blank ML et al (1979) Antihypertensive activity of an alkyl ether analog of phosphatidylcholine. Biochem Biophys Res Commun 90(4):1194–1200
Demopoulos CA, Pinckard RN, Hanahan DJ (1979) Platelet-activating factor. Evidence for 1-O-alkyl-2-acetyl-sn-glyceryl-3-phosphorylcholine as the active component (a new class of lipid chemical mediators). J Biol Chem 254(19):9355–9358
Benveniste J, Henson PM, Cochrane CG (1972) Leukocyte-dependent histamine release from rabbit platelets. The role of IgE, basophils, and a platelet-activating factor. J Exp Med 136(6):1356–1377
Marathe GK et al (1999) Inflammatory platelet-activating factor-like phospholipids in oxidized low density lipoproteins are fragmented alkyl phosphatidylcholines. J Biol Chem 274(40):28395–28404
Uemura Y, Lee TC, Snyder F (1991) A coenzyme A-independent transacylase is linked to the formation of platelet-activating factor (PAF) by generating the lyso-PAF intermediate in the remodeling pathway. J Biol Chem 266(13):8268–8272
Montrucchio G, Alloatti G, Camussi G (2000) Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev 80(4):1669–1699
Blank ML et al (1988) Stimulation of the de novo pathway for the biosynthesis of platelet-activating factor (PAF) via cytidylyltransferase activation in cells with minimal endogenous PAF production. J Biol Chem 263(12):5656–5661
Shindou H et al (2007) A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:LYSO-PAF acetyltransferase. J Biol Chem 282(9):6532–6539
Bito H et al (1994) Cloning, expression and tissue distribution of rat platelet-activating-factor-receptor cDNA. Eur J Biochem 221(1):211–218
Nakamura M et al (1991) Molecular cloning and expression of platelet-activating factor receptor from human leukocytes. J Biol Chem 266(30):20400–20405
Ye RD et al (1991) Characterization of a human cDNA that encodes a functional receptor for platelet activating factor. Biochem Biophys Res Commun 180(1):105–111
Brizzi MF et al (1999) Thrombopoietin stimulates endothelial cell motility and neoangiogenesis by a platelet-activating factor-dependent mechanism. Circ Res 84(7):785–796
Camussi G et al (1995) Platelet-activating factor directly stimulates in vitro migration of endothelial cells and promotes in vivo angiogenesis by a heparin-dependent mechanism. J Immunol 154(12):6492–6501
Camussi G et al (1997) Angiogenesis induced in vivo by hepatocyte growth factor is mediated by platelet-activating factor synthesis from macrophages. J Immunol 158(3):1302–1309
Montrucchio G et al (1994) Tumor necrosis factor alpha-induced angiogenesis depends on in situ platelet-activating factor biosynthesis. J Exp Med 180(1):377–382
Montrucchio G et al (1997) Nitric oxide mediates angiogenesis induced in vivo by platelet-activating factor and tumor necrosis factor-alpha. Am J Pathol 151(2):557–563
Montrucchio G et al (1998) Potential angiogenic role of platelet-activating factor in human breast cancer. Am J Pathol 153(5):1589–1596
Robert EG, Hunt JD (2001) Lipid messengers as targets for antiangiogenic therapy. Curr Pharm Des 7(16):1615–1626
Bussolino F et al (1995) Platelet activating factor produced in vitro by Kaposi’s sarcoma cells induces and sustains in vivo angiogenesis. J Clin Invest 96(2):940–952
Bussolati B et al (2000) PAF produced by human breast cancer cells promotes migration and proliferation of tumor cells and neo-angiogenesis. Am J Pathol 157(5):1713–1725
Axelrad TW et al (2004) Platelet-activating factor (PAF) induces activation of matrix metalloproteinase 2 activity and vascular endothelial cell invasion and migration. Faseb J 18(3):568–570
Deo DD et al (2002) Phosphorylation of STAT-3 in response to basic fibroblast growth factor occurs through a mechanism involving platelet-activating factor, JAK-2, and Src in human umbilical vein endothelial cells. Evidence for a dual kinase mechanism. J Biol Chem 277(24):21237–21245
Sirois MG, Edelman ER (1997) VEGF effect on vascular permeability is mediated by synthesis of platelet-activating factor. Am J Physiol 272(6 Pt 2):H2746–H2756
Rollin S et al (2004) VEGF-mediated endothelial P-selectin translocation: role of VEGF receptors and endogenous PAF synthesis. Blood 103(10):3789–3797
Maliba R et al (2006) Angiopoietins-1 and -2 are both capable of mediating endothelial PAF synthesis: intracellular signalling pathways. Cell Signal 18(11):1947–1957
Im SY et al (1996) Augmentation of tumor metastasis by platelet-activating factor. Cancer Res 56(11):2662–2665
Sato S et al (1999) Accelerated proliferation of epidermal keratinocytes by the transgenic expression of the platelet-activating factor receptor. Arch Dermatol Res 291(11):614–621
Ishii S et al (1997) Bronchial hyperreactivity, increased endotoxin lethality and melanocytic tumorigenesis in transgenic mice overexpressing platelet-activating factor receptor. Embo J 16(1):133–142
Barber LA, Spandau DF, Rathman SC, Murphy RC, Johnson CA, Kelley SW, Hurwitz SA, Travers JB (1998) Expression of the platelet-activating factor receptor results in enhanced ultraviolet B radiation-induced apoptosis in a human epidermal cell line. J Biol Chem 273(30):18891–18897
Sheng Y, Birkle DL (1995) Release of platelet activating factor (PAF) and eicosanoids in UVC-irradiated corneal stromal cells. Curr Eye Res 14(5):341–347
Calignano A et al (1988) Isolation and identification of platelet-activating factor in UV-irradiated guinea pig skin. J Pharmacol Methods 19(1):89–91
Travers JB et al (1995) Identification of functional platelet-activating factor receptors on human keratinocytes. J Invest Dermatol 105(6):816–823
De Fabo EC et al (2004) Ultraviolet B but not ultraviolet A radiation initiates melanoma. Cancer Res 64(18):6372–6376
Noonan FP et al (2001) Neonatal sunburn and melanoma in mice. Nature 413(6853):271–272
Zhang Q et al (2005) Staphylococcal lipoteichoic acid inhibits delayed-type hypersensitivity reactions via the platelet-activating factor receptor. J Clin Invest 115(10):2855–2861
Biancone L et al (2003) Platelet-activating factor inactivation by local expression of platelet-activating factor acetyl-hydrolase modifies tumor vascularization and growth. Clin Cancer Res 9(11):4214–4220
Fallani A et al (2006) Platelet-activating factor (PAF) is the effector of IFN gamma-stimulated invasiveness and motility in a B16 melanoma line. Prostaglandins Other Lipid Mediat 81(3–4):171–177
Ko HM et al (2007) Critical role for matrix metalloproteinase-9 in platelet-activating factor-induced experimental tumor metastasis. Int J Cancer 120(6):1277–1283
Mannori G et al (2000) Interaction of tumor cells with vascular endothelia: role of platelet-activating factor. Clin Exp Metastasis 18(1):89–96
Melnikova VO et al (2006) Platelet-activating factor mediates MMP-2 expression and activation via phosphorylation of cAMP-response element-binding protein and contributes to melanoma metastasis. J Biol Chem 281(5):2911–2922
Deo DD, Bazan NG, Hunt JD (2004) Activation of platelet-activating factor receptor-coupled G alpha q leads to stimulation of Src and focal adhesion kinase via two separate pathways in human umbilical vein endothelial cells. J Biol Chem 279(5):3497–3508
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Melnikova, V.O., Villares, G.J. & Bar-Eli, M. Emerging Roles of PAR-1 and PAFR in Melanoma Metastasis. Cancer Microenvironment 1, 103–111 (2008). https://doi.org/10.1007/s12307-008-0002-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-008-0002-7