
Abstract
The Committee on Gene Symbolization, Nomenclature and Linkage (CGSNL) of the Rice Genetics Cooperative has revised the gene nomenclature system for rice (Oryza) to take advantage of the completion of the rice genome sequence and the emergence of new methods for detecting, characterizing, and describing genes in the biological community. This paper outlines a set of standard procedures for describing genes based on DNA, RNA, and protein sequence information that have been annotated and mapped on the sequenced genome assemblies, as well as those determined by biochemical characterization and/or phenotype characterization by way of forward genetics. With these revisions, we enhance the potential for structural, functional, and evolutionary comparisons across organisms and seek to harmonize the rice gene nomenclature system with that of other model organisms. Newly identified rice genes can now be registered on-line at http://shigen.lab.nig.ac.jp/rice/oryzabase_submission/gene_nomenclature/.
Similar content being viewed by others
Introduction
The biological community is moving towards a universal system for the naming of genes. Emerging gene nomenclature systems have been described for a number of plants such as Arabidopsis thaliana [23], tomato [17], maize [13], and Medicago [25], as well as for Saccharomyces cereviseae [22] and for metazoans such as mouse [16] and humans [26]. The adoption of a common genetic language across diverse organisms is a great advantage for scientific communication and facilitates structural, functional, and evolutionary comparisons of genes and genetic variation among living things. With increasing emphasis on the molecular and biochemical nature of genes and gene products, it is important that the gene nomenclature system for rice (Oryza) reflect knowledge about the biochemical features of a specific gene, gene model, or gene family as well as about the phenotypic consequences of a particular allele in a given genetic background.
The current rules for gene names and gene symbols in rice are based on recommendations from the Committee on Gene Symbolization, Nomenclature and Linkage (CGSNL) of the Rice Genetics Cooperative [12]. Most of the early gene names and symbols are descriptive of visible phenotypes that provided the earliest evidence for the existence of a gene, and these names and symbols are widely used by the rice research community. With the completion of the rice genome sequence [7] and the emergence of new methods for detecting, characterizing, and describing genes, an expanded nomenclature system is needed that outlines a set of standard procedures for describing genes based on biochemical characterization and on DNA, RNA, and protein sequence analysis [27], in addition to the rules previously outlined for naming genes associated with phenotypic variants [12].
The focus of this publication is to summarize the rules for gene nomenclature in rice and, so far as possible, to harmonize the rice gene nomenclature system with that of other model organisms. We describe a set of rules for naming chromosomes and identifying loci, genes, and alleles based on biological function, mutant phenotype, and sequence identity, and suggest ways of dealing with aliases (synonyms), sequence variants, and loci identified by multiple annotations of the genome assemblies available from various sources. The nomenclature rules are based on the previous rice gene nomenclature system [12], but they have been expanded to accommodate sequence information based on the recommendations by members of the International Rice Genome Sequencing Project (IRGSP) as summarized at two Rice Annotation Project (RAP) meetings, namely RAP-1, held in Tsukuba, Japan in December 2004 and RAP-2, held in Manila, Philippines in December 2005. These rules have also been approved by the Sub-committee on CGSNL of the Rice Genetics Cooperative (http://www.shigen.nig.ac.jp/rice/oryzabase/rgn/office.jsp).
Though studies on rice genetics have been documented for over a century, the recent advances in large-scale mutagenesis experiments and sequencing of expressed sequence tags (ESTs), full-length cDNAs, and both the Oryza sativa ssp. japonica and O. sativa ssp. indica genomes of rice (O. sativa) have significantly added to our understanding of gene networks, gene function, and allelic and sequence diversity. Therefore, the nomenclature practice summarized in this report is designed to outline the rules for naming genes and alleles based on biological function and to facilitate the cross-referencing of gene annotations provided by multiple sequencing and annotation projects, namely, the IRGSP [7], RAP [20], The Institute of Genomic Research (TIGR) [30], Munich Information Center for Protein Sequences (MIPS) [11], National Center for Biotechnology Information (NCBI) [19], Syngenta [6], and Beijing Genomics Institute (BGI) [31] and to provide coherence for annotation of gene variants coming from the sequencing of different germplasm accessions [1, 15].
Results
Genome assemblies and systematic locus identifier (systematic_locus_ID)
A single rice species may support multiple genetic, physical, and sequence maps, gene annotations, and genome assemblies. Currently, the O. sativa genome is represented by the genome sequence of the O. sativa ssp. japonica cultivar, cv. Nipponbare, which was sequenced by the IRGSP (International Rice Genome Sequencing Project) [7], and by the O. sativa ssp. indica cultivar, cv. 93-11, which was sequenced by the BGI [28]. The Nipponbare sequence has been annotated by several groups, including RAP [8, 20], TIGR [29], NCBI-GenBank [19], MIPS [11], and Syngenta [6], while annotation of the O. sativa ssp. indica sequence, cv. 93-11, has been provided almost exclusively by the BGI [31]. In the case of Nipponbare, the same raw sequence generated by the IRGSP has been independently assembled and annotated by both RAP and TIGR, and thus, the rice community currently manages three independent genome assemblies (two for cv. Nipponbare and one for cv. 93-11) for the species O. sativa.
Each of these assemblies has an independently annotated set of loci representing gene models/transcription units anchored along pseudomolecules that differ in subtle ways from each other. A locus is defined as a position on the genome, and because each annotation group independently assigns locus identifiers (locus IDs) to all genes, transcripts, and proteins based on their position on the pseudomolecules, the same gene may have a different systematic_locus_ID, depending on the genome, the assembly, and the software used for annotation. Specifications of the rules used by each annotation group to assign systematic_locus_IDs for nuclear genes/transcripts/proteins, organellar genes/transcripts/proteins, and transposable elements are available on the RAP database [20], the TIGR Osa1 database [30], and the BGI-RIS [31]. Suggestions for assigning systematic_locus_IDs citing examples from the RAP database are provided towards the end of this article.
Annotated genes include protein-coding genes [open reading frames (ORFs)/CDSs], non-coding RNA genes [ribosomal RNA (rRNA), transfer RNA (tRNA), microRNA, small interfering (siRNA), small nucleolar RNA (snoRNA), small nuclear RNA (snRNA), etc.], and pseudogenes. The use of systematic_locus_IDs (described in detail later in this manuscript) provides a systematic approach to the assignment of gene identifiers, as well as easy recognition of the location of the locus on a sequenced rice genome. As a consequence, the locus ID can be used to identify and track a locus on a particular genome assembly and to establish an association between a gene model and a functionally characterized gene. Since a majority of sequenced/annotated genes currently have no known (experimentally confirmed) function, the systematic locus ID also provides a useful way of tracking information about putative gene function. As summarized in Table 1, genes may be classified based on computationally determined sequence similarity to a previously known gene (presumed homologue, orthologue, or paralogue), protein, or consensus feature (such as the functional domain of a protein). While sequence similarity alone is considered insufficient to warrant the assignment of a gene name, the information makes a critical contribution to the characterization of the gene. While systematic locus identifiers provide a unique nomenclature within a genome assembly and annotation data set, the methods used to assign locus_IDs typically differ slightly between annotation groups, and this, coupled with the differences in genome assemblies and in gene repertoire (i.e., between O. sativa ssp. japonica and O. sativa ssp. indica), make it difficult to definitively cross-reference genes and loci among assemblies. Thus, as the functions/phenotypes of genes are experimentally described, the CGSNL provides a unifying gene tracking system that is independent of the genome assembly and annotation version. As described below, each gene registered at CGSNL is uniquely identified by its gene full name and a gene symbol.
Registering genes in CGSNL’s database will facilitate the cross-referencing of genes among the multiple annotation systems and also between alleles and sequence variants. Genes with approved names and symbols will all be associated with a gene function or phenotype and, where possible, at the time of registration, researchers will be asked to identify a systematic locus identifier for the new gene from the RAP annotation database. Links to systematic_locus_IDs in other annotation databases (i.e., TIGR, BGI, etc.) will also be made where ever possible. Thus, at the time of registration, when a systemic locus identifier is provided to CGSNL, the version of the assembly and annotation must also be deposited to provide full documentation of the mappings. If available, a GenBank/DDBJ accession number should also be provided. This will help ensure accuracy and appropriate cross-referencing of information as illustrated in Table 2.
Accurate cross-referencing among rice pseudomolecules requires careful manual curation. Close paralogues, particularly when tandemly arrayed, and subtle differences in the structure of gene models across multiple assemblies of the same genome sequence present significant challenges. Researchers familiar with the particular characteristics of a new gene will be in the best position to provide accurate information about the gene and to ensure that the different rice genome annotations are progressively improved and updated.
Rules for chromosome names and gene symbolization in rice
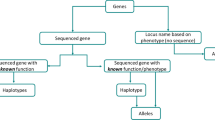
For purposes of this nomenclature system, a gene is defined as a segment of DNA that has a known or predicted function/phenotype. Sequenced genes that have no experimentally determined function/phenotype are not eligible for assignment of a gene name or gene symbol by the CGSNL (Fig. 1). Genes whose function/phenotype has been determined using classical genetics, but have not yet been associated with a sequence, are eligible for receiving a gene name and gene symbol, but may not have a systematic_locus_ID.

Schematic representation of genes receiving full names. “Genes” refers to the set of all genes in rice. “Sequenced genes” may be from a completely sequenced genome or other nucleotide sequence data sets. The subset of “sequenced genes of known function/phenotype” receives a gene name. Genes with “known function/phenotype, but without sequence” refers to genes that have no sequence information but do receive a gene name (based on their function/phenotype); often these are mapped on a genetic or physical map. “Sequenced genes of unknown function/phenotype” (this includes predicted genes, genes with full-length cDNA support, etc.) do not receive a name because they do not have experimental evidence supporting their function. However, various rice genome annotation projects provide systematic_locus_IDs that will serve as placeholders for names of these genes until they can be elevated into the category of sequenced genes of known function, at which time they will be assigned a gene name and a gene symbol.
Chromosome names
The 12 nuclear chromosomes are assigned Arabic numerals based on the convention outlined by Khush and Kinoshita [32], and linkage groups have been assigned to chromosomes and named accordingly. For database purposes, the chromosomes will each be assigned a two-digit number starting with 01 up to 12, but single digits for chromosomes 1–9 are generally used in publications. Short and long arms are symbolized by “S” and “L”, respectively (example: 1S, 1L), and it is acceptable to abbreviate them as chr. 1S and chr. 1L or Chr. 2S and Chr. 2L. While there are recognized inconsistencies in the current chromosome- and chromosome arm-naming conventions due to inaccuracies in the techniques previously used to estimate chromosome size and arm ratios [3], no revisions to the existing rice chromosome nomenclature have been suggested at this time. The circular chromosomes are assigned the English characters “Pt” for plastid or chloroplast, and “Mt” for mitochondria, respectively, instead of the Arabic numerals used for nuclear chromosomes. These chromosomes do not have centromeres, and thus, they will not be designated with short or long arms. It is acceptable to abbreviate them as chr. Pt or chr. Mt.
Gene full name
The full name of a gene consists of a name and a number referred to as the locus designator. Gene full names are written in all capital, italicized letters, with a space between the name and the locus number (i.e., SHATTERING 1). The name should briefly describe the salient characteristics associated with a biochemical function of the gene product or the phenotype rendered due to mutant or allelic forms of this gene. The locus designator consists of one to three digits and differentiates a gene at a particular locus from genes at other loci that confer a similar function or phenotype. The number used as the locus designator indicates the order in which a particular gene or gene family member was identified and should not be confused with the systematic_locus_ID or the chromosome/linkage group on which it is found. By default, any gene name that does not have a locus designator is presumed to be the first such gene identified and will be assigned the locus designator, “1”, e.g., PURPLE NODE will be designated PURPLE NODE 1. This format of writing the gene full name in all capital letters is different from the previous rule where the gene full name was written in all lowercase, italicized letters, with a capital first letter indicating dominant behavior and a lowercase first letter indicating recessive behavior of the first allele identified. Please refer to the section “Dominant/recessive relationships” for further discussion of this point.
In cases where a phenotype is mapped to a complex locus consisting of a tandem array of gene family members (for example, XANTHOMONAS ORYZAE PV. ORYZAE RESISTANCE 21, XA21, or SUBMERGENCE 1, SUB1), each gene in the array will be given an independent locus identifier (i.e., SUB1, SUB2, SUB3, etc.).
If a gene is newly identified based on sequence information and that gene is later proven to be the same as a gene originally identified based on phenotype (such as those listed by [12]), the precedence rule applies and the gene full name will be that based on phenotype, with the other name used as a synonym. If there is redundancy, overlap, or confusion caused by use of the same name for different genes or different names for the same gene, the first published gene name will generally be retained and the CSGNL will work with the authors of publications to identify a new gene name and gene symbol for the subsequently reported gene(s) or loci. Genes identified in the plastid genome will be assigned names and symbols as described by the Uniprot [24], and genes identified in the mitochondrial genome will be assigned names and symbols as recommended by [21].
Gene names are assigned based on experimental evidence about gene function or impact on phenotype. Experimental evidence may indicate a molecular function, a role in a biological process, or interaction with another gene or a phenotype associated with that gene (Fig. 1). Gene names based on computationally determined sequence similarity to a previously described homologue, orthologue, or paralogue, or based on the presence of a consensus feature such as an Interpro domain [18] can only be assigned if there is substantial experimental evidence confirming the gene’s function. Participants at the Rice Annotation Project-1 (RAP-1) meeting held at Tsukuba, Japan, in December 2004 agreed that database curators would use a standard system of ‘evidence categories’ to indicate the type of evidence or published experimental support for the nuclear gene annotation that they provide. A description of these categories is summarized in Table 1. As determined by CGSNL, if the evidence is considered insufficient to substantiate assigning a gene function, the gene name field remains empty and the description/definition field will be utilized to describe what is known about the characteristics of the gene (Table 1).
Gene symbol
The gene symbol is an abbreviation of the gene full name and the gene symbol is written in italics. A gene symbol consists of two parts, namely, a gene class symbol consisting of two to five letters, and the corresponding locus designator consisting of one to three digits. The gene symbol should be derived from the full name of the gene discussed previously, and it is followed by the same locus designator assigned to the full gene name. Both parts of the gene symbol should be written together with no space, hyphen, or any other symbol between them (e.g., SH1, GLH2). Together, the gene class symbol and locus designator form a gene symbol that must be unique to the locus and the genome. Every effort should be made to assign gene symbols that are easily recognizable as corresponding to a gene full name. Where possible, existing symbols should be retained even if they do not fully conform to this rule, for example: C (CHROMOGEN FOR ANTHOCYANIN), A (ANTHOCYANIN ACTIVATOR), and WX (GLUTINOUS ENDOSPERM). For any gene symbol that does not have a locus designator, it is presumed that the first such gene identified has the locus designator, “1”, e.g., the previously identified gene, GLUTINOUS ENDOSPERM (WX) should be designated GLUTINOUS ENDOSPERM 1 (WX1). All new genes with similar characteristics will be assigned a new number as the new locus designator by the CGSNL, in order of discovery. The CSGNL will also make sure that previously identified gene symbols and newly identified genes that were not previously registered are assigned a unique gene symbol, thus avoiding conflicting names and symbols.
The use of the suffix “(t)” and “*” to indicate a ‘tentative’ locus designation (when the allelic relationship between a newly described gene and a previously reported gene is not clear [12]) will be suspended and new genes will be assigned a new locus designation, under the assumption that they are new loci. If the new gene is later demonstrated to be allelic to a previously reported locus, the records of the two should be merged and the original gene symbol will be adopted by the precedence rule. The other symbol(s) will be cited as synonym(s). No previously assigned gene symbols will be deleted, thus avoiding confusion resulting from re-usage of the same symbol. Assigning a symbol to a gene should be consistent with that of the full gene name as described above.
Authors who refer to specific rice genes of known function in their publications must cite the approved gene full name and symbol, if available, a ‘systematic locus ID’ from one of the genome annotation centers and, if possible, a GenBank accession number. Where complete information is not yet available, either the systematic locus_ID or the gene symbol can be used as a placeholder until additional experimental evidence is provided (Fig. 1). Gene names must not be assigned unless approved by the CGSNL.
Use of species name in gene name and symbol
The use of organism-specific prefixes such as “Os” (O. sativa) in the gene name and/or gene symbol may be useful in publications but will not be included in the official gene name because it is redundant with species information that is already associated with submitted/registered genes. Furthermore, it leads to a proliferation of gene names Oryza sativa-X. The relationship between the gene and the organism will be clearly maintained in all genome and sequence databases. However, authors may append the organism-specific prefixes for clarity in publications to avoid repetition of the species name whenever a gene is referenced. In any case, the species symbol should not become part of the adopted gene symbol or gene full name. Note, however, that the symbol “Os” is allowed for use in the systematic locus ID, e.g., Os05g0000530, LOC_Os03g01590, and OsIBCD000082, that is assigned based on the system adopted by RAP (http://rapdb.lab.nig.ac.jp/index.html), TIGR (http://www.tigr.org/tdb/e2k1/osa1/tigr_gene_nomenclature.shtml), and BGI-RIS (http://rise.genomics.org.cn/rice/index2.jsp), respectively.
Allelic variants
Different alleles of the same gene are distinguished by adding a numerical suffix (or previously a letter), separated by a dash or hyphen, to the gene full name or the gene symbol, e.g., SHATTERING 1-1 (SH1-1); PGI1-1, PGI1-2. Historically, there are a few cases where a letter (t) or asterisk (*), rather than a number, was used to indicate an allele, and because these letter or symbol descriptions of allelic variants have become widely used and accepted in the rice genetics community, they will be retained as exceptions in publications and will be noted as synonyms in the database.
Dominant/recessive relationships
Historically, the gene full name was written in all lowercase, italicized letters, starting with an upper case letter if the first allele described in the literature was dominant, and with a lowercase letter if the first allele described was recessive. In view of recent advances in identifying genes based on sequence and large-scale genomics efforts, and the occurence of genes that are expressed only in haploid cell(s) (i.e., pollen or egg), the dominance or recessiveness of an allele or variant of a locus may be unknown or inconsequential. However, the dominance or recessiveness of an allele is still important to the genetics community when investigating gene function. Therefore, it is recommended that for publication purposes, and where a particular germplasm resource is being described, recessive alleles be indicated with all lower case letters and dominant alleles begin with an upper case first letter followed by lower case letters, with all letters in italics (similar to the previous convention; Table 3). Nonetheless, the official (generic) gene name will be written in all capital letters and the dominant/recessive behavior of particular alleles will be recorded as attributes of the alleles, rather than as part of the gene name in the database.
Sequence variants
Given that a gene is a DNA segment that has a known or predicted function/phenotype, once a gene has been named and located on a sequence map via a systematic locus_ID, it can also be represented by the group of alleles and sequence variants that consistently map to the same genetic locus. Molecular variants of genes identified by sequence alone in diverse plant material will be given a name, symbol, and accession identifier, and information about the sequence variant will be cross-referenced to specific information about the germplasm source (including the corresponding germplasm accession ID) from which the DNA/RNA material was isolated. However, sequence variants will not be considered “alleles” by the CGSNL until a molecular function or phenotype has been described for them and an allelism test has been performed. “Sequence variants” whose specific function is unknown will be distinguished from “alleles” by adding a suffix ‘-sX, to an allele name, where “s” means “sequenced” and “X” is a number that serves to identify a particular sequence variant. The name and symbol of a molecular variant will carry the name and symbol of the corresponding gene, similar to the convention for an allele, except that it carries the suffix described above and is written in all caps due to the fact that no allelic behavior can be assigned to these sequenced variants (Table 3).
If a sequenced variant is later demonstrated to confer a specific novel phenotype or function, it will be assigned a new allele identifier or alternatively, if a sequenced variant is demonstrated to be equivalent to a previously named allele corresponding to a known gene, it will be assigned an existing allele identifier, based on the precedence rule, with the other identifier retained as a synonym. An example of recommended designation of gene locus, full name, and allele is shown in Table 3. The germplasm name and its accession information, in which sequence variants are identified, are not recorded in the official name/symbol. This information should be recorded separately in the database so that it can be readily cross-referenced by the genetics community. Authors submitting information about sequence variants will be responsible for finding out if the newly sequenced form is the same as any previously reported sequence variant or allele. In publications, authors may choose to concatenate the allele name, sequence variant suffix, and the germplasm source to avoid undue repetition for the readers.
Protein name and symbol
The name of a protein encoded by a particular gene should be consistent with the gene full name in cases where the gene name is based on phenotype or molecular function (refer to the “Gene full name” section), except that the protein name is written using all upper case characters without italics. If, at a later stage, a gene and its corresponding protein product are determined to have a biochemically characterized molecular function, such as an enzyme or a structural component (subunit) of a macromolecular complex, the protein should be assigned a synonym consistent with the enzyme nomenclature recommended by the IUPAC Enzyme Commission or the macromolecule name adapted by the IUBMB [4]. Because there may be several functional assignments for a given protein (i.e., based on a phenotypic assay, a biochemical assay, or a molecular function), there may be several synonyms for the protein name (and similarly, for the gene full name). The protein symbol should always be consistent with the adopted gene symbol, with the exception that protein symbols are written using all upper case characters without italics, followed by a space and the numeric locus designator. For example, the GLUTINOUS ENDOSPERM 1 (WX1) gene encodes the granule-bound starch synthase enzyme (EC: 2.4.1.11). The protein name is GLUTINOUS ENDOSPERM 1 and the symbol is ‘WX1’. The protein name(s), ‘WAXY’, ‘WAXY 1’, and GRANULE-BOUND STARCH SYNTHASE (GBSS) will be recorded as synonyms. If a name cannot be assigned based on phenotype, known biochemistry, or other experimental evidence supporting its function, a systematic locus identifier (described above) and a name consistent with the description in Table 1 must be used to describe the gene until its function can be confirmed.
Post-translational modification
In cases where a post-translational modification, such as protein splicing, leads to formation of two or more protein molecules with different activities or functions, the spliced protein molecules will carry a protein name and symbol consistent with their molecular function or associated phenotype, and will carry the name and symbol from the primary molecule as synonyms.
Pseudogenes
Molecular technology has identified sequences that bear striking similarity to structural gene sequences but are not transcribed. These sequences are termed pseudogenes. In order to show the relatedness of pseudogenes to functional genes, pseudogenes will be identified with the gene symbol of the structural/functional gene, in italics, followed by a “.P” (symbol “period” and capital letter “P”) for pseudogene. This will replace the conventionally used Greek symbol for “psi” for pseudogene; an example is RPS14.P instead of RPS14.psi for pseudoribosomal protein S14. The same is suggested for pseudogenes identified in mitochondrial and plastid (chloroplast) genomes and examples are ACTB.P1 (ACTIN BETA PSEUDOGENE 1), ACTB.P2 (ACTIN BETA PSEUDOGENE 2), etc. Pseudogenes may be on different chromosomes or closely linked to the functional gene from which they derive their name and may occur in varying numbers. For nomenclature purposes, a pseudogene is a gene that has no function [5]. If a pseudogene were later proven to transcribe and regulate the expression of another gene or for instance the transcribed mRNA were shown to have a function, the gene would have to be reclassified to another gene category such as fnRNA or potogene as described by [2].
Unmapped genes
Due to the genetic variability inherent within a species, it is possible that a gene sequenced from one germplasm accession may not be mapped in either of the two fully sequenced genomes from O. sativa, due to insertion/deletion polymorphism and gene family expansion/contraction. Similarly, a gene identified by phenotype in a segregating population may not be present in one of the parental genomes. In such situations, even without the mapping information, a gene name and symbol can still be assigned to these allelic variants. When assigning a gene name to such unmapped loci, it is essential to confirm that there is valid experimental evidence supporting the existence and function of the gene. If a second instance of a similar unmapped sequenced gene occurs, the best reciprocal match approach should be applied to rigorously confirm whether it is, in fact, the same as the gene previously identified. In cases where a second instance of a phenotypically defined gene occurs, an allelism or complementation test will be considered essential evidence. If any of these evidences are missing, such a gene should be assigned a new gene name and symbol. In the mean time, the unique identifier assigned for a gene that is registered by the CGSNL, and if available the GenBank accession number, will serve as a placeholder.
Quantitative trait loci (QTL)
QTLs serve as placeholders for genes and contribute to the functional characterization of the genome. A QTL is defined as a region of the genome that is statistically associated with a measurable phenotype, generally with a quantitatively inherited trait. QTLs are identified by genetic mapping using association panels of segregating populations, and each QTL is defined by at least two, closely linked, mapped genetic markers that delimit a specific chromosomal region.
Rice QTL nomenclature rules [14] indicate that each QTL name should be italicized and start with a lower case letter “q” to indicate that it is a QTL, followed by a two to five letter standardized “trait name” (e.g. SW for Seed Width), a number designating the rice chromosome on which it occurs (1–12), a period (“.”), and a unique identifier to differentiate individual QTLs for the same trait that reside on the same chromosome (e.g. qSW5.1). When QTLs are entered into a genome database such as Gramene [9], they may be further assigned a standardized trait term from the Trait Ontology (TO; [10]; e.g. seed width, Accession #TO: 0000140) to facilitate querying and may be assigned a new, unique identifier to avoid confusion between studies. In any case, this database assignment will be reflected as a synonym within the QTL record, and the original, published QTL name will be retained for search purposes.
When gene(s) that are actually responsible for the phenotypic variation associated with the QTL is identified for the first time based on its correspondence to a QTL, the gene full name may reflect the QTL designation (except for the elimination of the prefix ‘q’ and the use of italics (e.g., SW5)); however, if the gene underlying the QTL corresponds to a previously characterized and named gene, the precedence rule applies and the original gene name must be retained. Nonetheless, it is recommended that the relationship between genes and QTLs be noted in the list of synonyms associated with gene names.
Systematic locus ID assignment: a RAP database example
Systematic locus ID for nuclear genes
Systematic locus identifiers will be assigned to genes identified along the rice (O. sativa ssp. japonica, cv. Nipponbare) pseudomolecules (assembled chromosome contigs of the sequenced genome of O. sativa) based on automated gene prediction programs, orthologue alignments, and/or alignment of ESTs and full-length cDNAs, following the recommendations adopted for yeast S. cereviseae [22] and A. thaliana [23]. Systematic identifiers are assigned to protein-coding genes (ORFs), RNA-coding genes (snoRNA, snRNA, rRNA, tRNAs, and microRNAs), and pseudogenes. A nuclear gene locus ID will consist of: (a) an uppercase letter “O” and lowercase letter “s” to indicate the rice species O. sativa; (b) a two-digit number to indicate a specific rice chromosome (01, 02, 03, ...12); (c) a letter “g” indicating that the locus ID is for a gene; (d) a seven-digit number (assuming there will be fewer than 10,000 genes per chromosome) indicating the sequential order of a gene along a chromosome in ascending order from the telomere of the short arm (north side) to the telomere of the long arm (south side). The numbers indicating gene order are independent of the polarity of the strand (+/– or Watson/Crick) and should be initially assigned in increments of 100, thus leaving room for expansion as new genes are discovered. For example, the third and fourth genes on rice chromosome 5 would be indicated as Os05g0000300 and Os05g0000400.
If, during the course of the sequencing or based on new experimental evidence, a new gene is detected between the two already annotated genes, the new gene will be assigned a number between the two previously annotated genes, using the tenth number space. For example, a gene discovered between Os05g0000300 and Os05g0000400 would be assigned Os05g0000350, again leaving room for expansion. Despite the obvious benefits of this strategy, it is true that in some cases gene order within a particular chromosomal segment may not follow the ascending/descending order rule based on precedence of gene discovery; however, this shortcoming does not negate the value of the system as a whole. Systematic locus IDs will be assigned to all genes, including those that are known to have been introduced into the nuclear genome via an insertion of a portion of an organellar genome (plastid and/or mitochondria), recognizing that such genes will often turn out to be non-functional or pseudogenes.
For regions where the genome sequence of rice is incomplete, such as the gaps in the telomeric and centromeric regions or the smaller interstitial gaps, it is suggested that a locus ID space be reserved. The locus ID space would accommodate 1,000 genes per gap in the telomere and centromere regions, and one gene per 2 kb interstitial gap.
Note that the loci identified in the genomes of cultivars, subspecies, or species accessions of the genus Oryza other than O. sativa ssp. japonica cv. Nipponbare must be named in consultation with the CGSNL. Database curators and individual researchers must assign names and symbols only after registration with and approval by the CGSNL.
Systematic locus ID for organellar genes
The main mitochondrial and chloroplast chromosomes are circular (also called master circles) and do not have arms. Locus IDs for genes found on organellar chromosomes will use the symbols ‘Mt’ for mitochondrion and ‘Pt’ for plastid (chloroplast), respectively, instead of the chromosome number designations used for nuclear genes. These letters will be followed by a letter “g” indicating that the locus corresponds to a gene, followed by a seven-digit number (assuming there will be fewer than 10,000 genes per chromosome) indicating the sequential order of genes along an organellar chromosome, independent of the polarity of the strand, in ascending order from the first base pair of the completely sequenced molecule to the last base pair in the linearized molecule (as submitted by the author of the sequence to any of the reference sequence databases, namely NCBI-GenBank, DDBJ, or EMBL). For example, OsPtg0000100 indicates the first gene on the rice plastid genome. Looking at the GenBank entries for plastid genomes sequenced from O. sativa cv. Nipponbare, this would refer to the gene, PSBA (82–1,143 bp), as referenced by GenBank entry NC_001320.
In addition to the system for identifying loci found on master circles, there are genes found on plasmids, both linear and circular (also referred to as subgenomic circles) in the mitochondria, and these will be indicated by using a lower case letter, a–z (in the order of precedence by submission to GenBank), immediately following the organellar symbol, Mt or Pt. For example, OsMtag0000200 indicates gene 2 on the 2,135-bp mitochondrial plasmid B1 (GenBank accession NC_001751). The number series for genes on plasmids will start from the first base pair of the fully assembled, sequenced plasmid or subgenomic circle, as determined by the sequence submitted to GenBank, DDBJ, or EMBL.
Transcript ID
Every known or predicted form of transcript of a gene will be assigned a systematic identifier that will be the same as the locus identifier except that the letter ‘g’ for gene will be replaced by letter ‘t’ for transcript, to be added as a suffix following the two-digit chromosome identifier. This naming convention will ensure consistency in the gene’s locus ID and its transcript ID. For example, the transcript Os05t0000300 is transcribed by the locus Os05g0000300 representing gene 3 on chromosome 5. Sometimes the nascent transcript undergoes alternative splicing. In order to clearly identify the alternatively spliced forms of the transcripts, a two-digit suffix will be added to the systematic transcript ID of the gene will be added, separated by a dash, e.g., -01, -02, -03, ....-99, in order of discovery. By default, the transcript ID of the very first transcript (or the only transcript identified) will always have number “-01” suffixed to the transcript ID. For example, the transcript ID of the locus Os05g0000300, for which there are no known splice variants, will be Os05t0000300-01. If there is a later report suggesting that the transcript from this locus undergoes alternative splicing, such that three alternative forms are created, if any one of the three forms matches the original transcript, it would retain the original transcript ID and two additional IDs would be generated, Os05t0000300-02 and Os05t0000300-03. Assigning the number series to the splice variants will depend on the precedence of identification, the submission to GenBank or possibly the size of the cDNA. Any additional alternative forms are numbered sequentially.
Protein ID
All the peptides deduced experimentally or computationally from a gene sequence/transcript will be assigned a systematic identifier that is the same as the transcript identifier, except that the letter ‘t’ for transcript will be replaced by the letter ‘p’ for protein, thus assuring consistency with the gene’s locus ID and its transcript ID. For instance, the protein Os05p0000300-01 is translated from transcript Os05t0000300-01 that is transcribed from locus Os05g0000300, which represents gene 3 on chromosome 5. In order to avoid conflicts with proteins deduced from alternatively spliced forms of the transcripts from a single locus, the protein ID must reflect the corresponding transcript from which it is deduced, except for the letter ‘t’.
Genes present on unanchored sequenced clones
For genes identified in unanchored BAC/PAC clones, continued use of the nomenclature system whereby the gene is sequentially designated by a numerical suffix following the BAC/PAC clone name assigned by the sequencing center (e.g., F23H14.13) is acceptable. The systematic locus ID nomenclature system outlined above will supersede the clone-based name once the sequence in the region is fully assembled and completed. In such cases, the earlier clone-based locus identifiers must become either the alternate ID or the gene synonym.
Adding, deleting, editing, merging, and splitting of loci
Editing a locus
Consistent use of a given locus identifier, full gene name, and gene symbol is suggested. Consistency can be maintained as long as there are no major changes in the gene model or function, particularly no changes that would lead to a change in the start position of the locus. For example, consistency of nomenclature is possible in cases where the gene encodes an ORF, and the modifications in annotation change only the intron–exon boundaries, the strand identity, require the addition or deletion of exon(s) or intron(s), or change or modify the function or associated phenotype assigned to the locus. Similarly, in cases where updated annotation changes the definition of the ORF, the gene’s full name, symbol, and the definition line of the GenBank/DDBJ/EMBL records should reflect the change in the molecule’s structure or function, but in all of the above cases, the locus ID remains same.
Deleting a locus
Genes identified by computational methods alone may prove to be false positives when confirmed by experimental evidence, thus making it necessary to retire the locus. In such cases, all the records and corresponding identifiers should be preserved with a flag OBSOLETE and never DELETED from data repositories. The flag OBSOLETE ensures that the same identifiers are not used again for a new locus, thus avoiding a situation that would lead to confusion and, if required, makes it possible for an obsolete gene to still be referenced.
Splitting a locus
When it is determined that a locus identifier actually refers to more than one gene (e.g., two genes mistakenly identified as one by an automated prediction method), the locus closest to the locus start position will retain the original locus identifier, gene name, and gene symbol, and the gene farther from the locus start position will be considered a newly identified locus and will receive a new locus identifier, gene name, and gene symbol, following the recommendations mentioned above. The modification of the gene name and gene symbol should accommodate the new function, if applicable.
Merging loci
In the cases where there is experimental evidence (such as full-length cDNA sequence) indicating that two previously identified genes are actually one gene or part of the same locus, the two loci must be merged into one. The new locus must retain the locus identifiers, gene name, and gene symbol from the locus closest to the start position of the new, merged locus. For the second gene, the locus identifier becomes a secondary locus ID (associated with the first one), whereby the second gene’s name and symbol will become synonyms of the first one.
Transposable element locus ID
IDs assigned to loci containing a transposable element (TE) will be similar to those for gene loci except that the ‘g’ in the gene locus ID will be replaced by ‘te’, e.g., Os05te0000300. Since the majority of the current TE annotations are based on in silico prediction and computational analyses, it was decided at the RAP1 meeting that this system be implemented at a later stage. It was also suggested that experts be consulted before a nomenclature system for TEs is put in place. However, if a TE is proven to contain a functional gene, it will be assigned a gene locus identifier, as described above.
Registration of gene names and symbols
A web-based gene registration and nomenclature website has been established to support the registration process and can be accessed at http://shigen.lab.nig.ac.jp/rice/oryzabase_submission/gene_nomenclature/. Registration requests will be handled by subcommittees within the CGSNL, depending whether the gene was identified by sequence or by phenotype. Rice researchers are encouraged to use this website to register genes and alleles of interest. The CGSNL will give priority to functionally characterized genes and may request experimental evidence in order to process a new request. The approved gene names and symbols will be released immediately upon approval. Although this nomenclature system will catalogue the genes from O. sativa, every effort will be made by the CGSNL to manage gene nomenclature in non-O. sativa rice species and the rice community is encouraged to use the same gene registration site for registering rice genes from species other than O. sativa.
Registration process
The following types of information should be submitted when registering a new gene:
-
1.
Descriptive information about the characteristics of the gene, including but not limited to information about its molecular function, its role in a biological process, its location in a subcellular component, its expression in a particular plant tissue and growth stage, and its effects on phenotype
-
2.
Inheritance and allelism data
-
3.
Source germplasm (genus, species, stock/strain/Accession_ID/germplasm repository). If from a hybrid accession, provide information on germplasm resources of the parents
-
4.
Chromosomal and map location
-
5.
Sequence data and gene model (intron/exon structure, promoter, etc.)
-
6.
GenBank accession number and/or locus_ID from at least one of the rice genome annotation projects (if available)
-
7.
Protein/gene family relationship
-
8.
Supporting documents including a photograph of the mutant phenotype, RNA and/or protein expression data, enzymatic assays, sequence alignments, etc.
The submitted registration entries will be sent to the convener of the CGSNL via an electronic submission form provided via the OryzaBase database that will host the gene registration site (http://shigen.lab.nig.ac.jp/rice/oryzabase_submission/gene_nomenclature/). After examining the submitted information to determine if a gene is new and to consider naming conventions, the convenor will notify the submitting author to verify the new gene’s full name and symbol. Upon approval, the registered gene will be assigned an appropriate gene full name and gene symbol. This must be reported in the annotation databases and in publications. The gene registration database will also provide an online and downloadable list of registered genes that will include information on the approved gene name, symbol, synonyms, mapped systematic_locus_IDs from annotation databases, and the associated GenBank accessions, if available (Table 2). The convenor must also communicate with the appropriate databases and RGC members so that the new gene name and symbol is included in the list of genes/alleles published in the OryzaBase (http://www.shigen.nig.ac.jp/rice/oryzabase/top/top.jsp), Gramene (http://www.gramene.org), IRIS (http://www.iris.irri.org), RAP (http://rapdb.lab.nig.ac.jp/), TIGR (http://www.tigr.org/tdb/e2k1/osa1/), and other relevant databases and websites. A research note describing all newly accepted genes/alleles will be published biannually in the Rice Genetics Newsletter (http://www.shigen.nig.ac.jp/rice/oryzabase/rgn/newsletter.jsp).
Amendments
Suggestions for amendments of these rules can be submitted to the CGSNL using an online “Suggestions” form available on the OryzaBase web site http://shigen.lab.nig.ac.jp/rice/oryzabase_submission/gene_nomenclature/. Amendments will be announced in the journal RICE, in the Rice Genetics Newsletter and via the OryzaBase, Gramene, and IRIS Databases and the rice-e-net e-mail list (http://chanko.lab.nig.ac.jp/list-touroku/rice-e-net-touroku.html). For contact, the users are encouraged to send e-mail to genenomenclature@chanko.lab.nig.ac.jp.
Discussion
By curating genes whose function has been experimentally determined (‘genes of known function’) independently of genes predicted by sequence analysis alone (gene models), the rice community has established a flexible yet robust system for bridging these two different approaches to gene structure/function analysis. The long-term goal is to provide a functional description for every gene in rice, at which time, every gene model (locus_ID) should be associated with a gene name. However, with the rapidly diminishing cost of sequencing and the rapidly expanding number of sequenced rice genomes in the public domain, our understanding of the gene repertoire in rice is no longer limited by the availability of a single O. sativa ssp. japonica and a single O. sativa ssp. indica genome sequence. Thus, the rice gene nomenclature system has adopted protocols for establishing one-to-many associations between genes of known function and computationally determined gene models, where multiple types of evidence are curated in support of the functional description of each Oryza gene.
A gene may code for a protein product (CDS) or it may code for one of many kinds of non-coding RNA molecules, including snoRNA, snRNA, tRNA, rRNA, microRNA, SiRNA, or fnRNA (functional RNA), etc. If new classes of genes are identified in the future, we will amend our classification system accordingly.
In the naming of genes, the use of English is preferred, and gene symbols should consist of Latin letters and Arabic numerals. The name of a gene should either briefly describe the phenotype and/or convey some meaning as to the function of the gene product, if known. All new gene names should be approved by and registered with the CGSNL to avoid confusion and duplication. The rice community gives priority to the first published name for a gene but it is recognized that names change over time to reflect new knowledge. While we do not propose the adoption of a rigid or restrictive gene nomenclature system at this time, we agree to adopt a system of synonyms that permits the establishment of correspondences between sequence-based gene identifiers and names based on experimentally confirmed biochemical function or phenotypic variation. This approach allows for continued evolution of the gene nomenclature system for rice as new technologies are developed and new knowledge is accumulated.
References
Ammiraju JSS, Luo M, Goicoechea JL, Wang W, Kudrna D, Mueller C, et al. The Oryza bacterial artificial chromosome library resource: construction and analysis of 12 deep-coverage large-insert BAC libraries that represent the 10 genome types of the genus Oryza. Genome Res 2006;16:140–7.
Brosius J, Gould SJ. On “genomenclature”: a comprehensive (and respectful) taxonomy for pseudogenes and other “junk DNA”. Proc Natl Acad Sci USA 1992;89:10706–10.
Cheng Z, Buell CR, Wing RA, Gu M, Jiang J. Toward a Cytological Characterization of the Rice Genome. Genome Res 2001;11:2133–41.
Committees, Biochemical Nomenclature (2006) IUPAC-IUBMB Joint Commission on Biochemical Nomenclature (JCBN). 2006. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB).
Gerstein M. Pseudogene.org. http://pseudogene.org/main.html. 2006, Accessed March 25, 2008.
Goff SA, Ricke D, Lan T-H, Presting G, Wang R, Dunn M, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002;296:92–100.
IRGSP. The map-based sequence of the rice genome. Nature 2005;436:793–800.
Itoh T, Tanaka T, Barrero RA, Yamasaki C, Fujii Y, Hilton PB, et al. Curated genome annotation of Oryza sativa ssp. japonica and comparative genome analysis with Arabidopsis thaliana. Genome Res 2007;17:175–83.
Jaiswal P, Ni J, Yap I, Ware D, Spooner W, Youens-Clark K, et al. Gramene: a bird’s eye view of cereal genomes. Nucleic Acids Res 2006;34:D717–23.
Jaiswal P, Ware D, Ni J, Chang K, Zhao W, Schmidt S, et al. Gramene: development and integration of trait and gene ontologies for rice. Compar Funct Genom 2002;3:132–6.
Karlowski WM, Schoof H, Janakiraman V, Stuempflen V, Mayer KFX. MOsDB: an integrated information resource for rice genomics. Nucleic Acids Res 2003;31:190–2.
Kinoshita T. Report of the committee on gene symbolization, nomenclature and linkage groups. Rice Genet Newslett 1986;3:4–8.
MaizeGDB. A Standard For Maize Genetics Nomenclature. http://www.maizegdb.org/maize_nomenclature.php. 2002, Accessed 10 April 2008.
McCouch SR, Cho YG, Yano M, Paul E, Blinstrub M, Morishima H, et al. Report on QTL nomenclature. Rice Genet Newslett 1997;14:11.
McNally KL, Bruskiewich R, Mackill D, Buell CR, Leach JE, Leung H. Sequencing multiple and diverse rice varieties. Connecting whole-genome variation with phenotypes. Plant Physiol 2006;141:26–31.
MGNC, Mouse Genomic Nomenclature Committee. Mouse Nomenclature Home Page http://www.informatics.jax.org/mgihome/nomen/. 2005, Accessed 10 April 2008.
Mueller L. SOL Project Sequencing and Bioinformatics Standards and Guidelines. http://www.sgn.cornell.edu/documents/solanaceae-project/docs/tomato-standards.pdf. 2005, Accessed 10 April 2008.
Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. InterPro, progress and status in 2005. Nucleic Acids Res 2005;33:D201–5.
NCBI. Entrez genomes: Oryza sativa (rice) genome. http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?taxid=4530. 2006, 10 April 2008.
Ohyanagi H, Tanaka T, Sakai H, Shigemoto Y, Yamaguchi K, Habara T, et al. The Rice Annotation Project Database (RAP-DB): hub for Oryza sativa ssp. japonica genome information. Nucleic Acids Res 2006;34:D741–4.
Price CA, Reardon EM, Lonsdale DM. A guide to naming sequenced plant genes. Plant Mol Biol 1996;30:225–7.
SGD. SGD Gene Naming Guidelines. http://www.yeastgenome.org/gene_guidelines.shtml. 2005, 10 April 2008.
TAIR. Arabidopsis Nomenclature. http://www.arabidopsis.org/info/guidelines.jsp. 2005, 10 April 2008.
UniProt. List of plastid encoded proteins. http://ca.expasy.org/cgi-bin/lists?plastid.txt. 2006, 10 April 2008.
VandenBosch KA, Frugoli J. Guidelines for genetic nomenclature and community governance for the model legume Medicago truncatula. Mol Plant-Microb Interact 2001;14:1364–7.
Wain HM, Lush MJ, Ducluzeau F, Khodiyar VK, Povey S, et al. Genew: the Human Gene Nomenclature Database, 2004 updates. Nucleic Acids Res 2004;32:D255–7.
Wu R, Hirai A, Mundy J, Nelson R, Rodriguez R. Guidelines for nomenclature of cloned genes or DNA fragments in rice. Rice Genet Newslett 1991;8:51–3.
Yu J, Hu S, Wang J, Wong GK-S, Li S, Liu B, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002;296:79–92.
Yuan Q, Ouyang S, Liu J, Suh B, Cheung F, Sultana R, et al. The TIGR rice genome annotation resource: annotating the rice genome and creating resources for plant biologists. Nucleic Acids Res 2003;31:229–33.
Yuan Q, Ouyang S, Wang A, Zhu W, Maiti R, Lin H, et al. The Institute for Genomic Research Osa1 rice genome annotation database. Plant Physiol 2005;138:18–26.
Zhao W, Wang J, He X, Huang X, Jiao Y, Dai M, et al. BGI-RIS: an integrated information resource and comparative analysis workbench for rice genomics. Nucleic Acids Res 2004;32:D377–82.
Khush GS, Kinoshita T. Rice karyotype, marker genes, and linkage groups. In: Khush GS, Toenniessen GH, editors. Rice biotechnology. Wallingford, Oxon, UK and Manila, Philippines: CAB International and IRRI; 1991. p. 83–108.
Acknowledgments
We kindly acknowledge the following researchers, Pankaj Jaiswal, Junjian Ni, and Immanuel Yap from the Gramene database (http://www.gramene.org) and the Department of Plant Breeding and Genetics at Cornell University, Ithaca, NY, USA; Toshiro Kinoshita from the Kita 6 Jo, Nishi 18 Chome, Sapporo 060-0006, Japan; David Mackill and Richard Bruskiewich from the International Rice Research Institute, DAPO 7777, Metro Manila, Philippines; C. Robin Buell from Department of Plant Biology, Michigan State University, East Lansing, MI 48824-1312, USA; Masahiro Yano, Takeshi Itoh, and Takuji Sasaki from the Department of Molecular Genetics, National Institute of Agrobiological Resources, Tsukuba, Ibaraki 305-8602, Japan; and Qifa Zhang from the National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, 430070, People’s Republic of China for the help in preparing this manuscript and to numerous other experts for their help and useful suggestions on improving the rice gene nomenclature. We also thank all the members of the Rice Genetics Cooperative (RGC: http://www.shigen.nig.ac.jp/rice/oryzabase/rgn/office.jsp) for their support. Financial support was provided by NSF Grant DBI 0703908 (Cold Spring Harbor Subcontract 22930113 to Cornell University).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
The current ex-officio member list below is correct as of the date of this galley proof. The current ex-officio members of CGSNL (The Committee on Gene Symbolization, Nomenclature and Linkage) are:
Atsushi Yoshimura from the Faculty of Agriculture, Kyushu University, Fukuoka, 812-8581, Japan, email: ayoshi@agr.kyushu-u.ac.jp
Guozhen Liu from the Beijing Institute of Genomics of the Chinese Academy of Sciences, Beijing, People’s Republic of China, email: gzhliu@genomics.org.cn
Yasuo Nagato from the Graduate School of Agricultural and Life Sciences, University of Tokyo, Tokyo 113-8657, Japan, email: anagato@mail.ecc.u-tokyo.ac.jp
Susan McCouch from the Department of Plant Breeding & Genetics, Cornell University, Ithaca, NY 14853-1901, email: SRM4@cornell.edu.
Yukiko Yamazaki from the National Institute of Genetics, Mishima, 411-8580, Japan, email: yyamazak@lab.nig.ac.jp
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
McCouch, S.R., CGSNL (Committee on Gene Symbolization, Nomenclature and Linkage, Rice Genetics Cooperative). Gene Nomenclature System for Rice. Rice 1, 72–84 (2008). https://doi.org/10.1007/s12284-008-9004-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12284-008-9004-9