
Abstract
Norovirus (NoV) is the most common viral cause of acute gastroenteritis worldwide. Vitamin A has demonstrated the potential to protect against gastrointestinal infections. However, the effects of vitamin A on human norovirus (HuNoV) infections remain poorly understood. This study aimed to investigate how vitamin A administration affects NoV replication. We demonstrated that treatment with retinol or retinoic acid (RA) inhibited NoV replication in vitro based on their effects on HuNoV replicon-bearing cells and murine norovirus-1 (MNV-1) replication in murine cells. MNV replication in vitro showed significant transcriptomic changes, which were partially reversed by retinol treatment. RNAi knockdown of CCL6, a chemokine gene that was downregulated by MNV infection but upregulated by retinol administration, resulted in increased MNV replication in vitro. This suggested a role of CCL6 in the host response to MNV infections. Similar gene expression patterns were observed in the murine intestine after oral administration of RA and/or MNV-1.CW1. CCL6 directly decreased HuNoV replication in HG23 cells, and might indirectly regulate the immune response against NoV infection. Finally, relative replication levels of MNV-1.CW1 and MNV-1.CR6 were significantly increased in CCL6 knockout RAW 264.7 cells. This study is the first to comprehensively profile transcriptomes in response to NoV infection and vitamin A treatment in vitro, and thus may provide new insights into dietary prophylaxis and NoV infections.
Similar content being viewed by others

Introduction
Norovirus (NoV) belongs to the Caliciviridae family along with Sapovirus, Lagovirus, Vesivirus, and Nevovirus (Vinje et al., 2019). It is a single-stranded, non-enveloped, positive-sense RNA virus, and is the most common cause of acute viral gastroenteritis. NoV-associated acute gastroenteritis is often fatal in children and the elderly, causing significant morbidity and mortality in these age groups (Patel et al., 2008). NoV is highly contagious and typically transmitted via direct contact, food, water, or fomites (Lindesmith et al., 2003). NoV infection causes various clinical symptoms, including diarrhea, nausea, vomiting, abdominal pain, and fever that lasts for 1–3 days. Unfortunately, there is no effective treatment or vaccine for NoV infection.
The murine NoV (MNV) model using murine macrophage cell line RAW 264.7 provides effective inhibition using physicochemical agents, such as TiO2, UV light, and pulsed light (Lee et al., 2008; Vimont et al., 2015). Anti-NoV effects of nucleoside analogues and the effect of simvastatin, a HMG-CoA reductase inhibitor, on NoV replication have been characterized in NV replicon-harboring cells (HG23 cells) (Chang, 2009).
Previous studies have demonstrated that interferon (IFN)-α reduces NoV infectivity (Chang, 2009), and that IFN-λ controls persistent enteric MNV infections (Nice et al., 2015). Mice lacking either type I or type II IFN (IFN-α/-β/-γ) or STAT-1 had a defect in immune responses of MNV infections (Karst et al., 2003; Mumphrey et al., 2007). In addition, Toll-like receptor-3 (TLR-3) and melanoma differentiation-associated protein-5 (MDA-5) are important for immune recognition of MNV (McCartney et al., 2008).
Retinoic acid (RA), a metabolite of dietary vitamin A, contributes to both innate and adaptive immune responses (Hall et al., 2011). Recent evidence indicates that loss of RA impairs immunity, whereas excess expression can induce inflammatory disorders (Hall et al., 2011). Retinoic acid-inducible gene I (RIG-I) signaling plays a crucial role in antiviral responses to viral RNA by producing type-I IFNs (Takeuchi & Akira, 2009). A recent study reported that sufficient vitamin A supplementation reduced mortality from bacterial, viral gastrointestinal and respiratory infections (Thornton et al., 2014). Despite knowledge of the essential role of vitamin A, its deficiency remains a global public health concern. An estimated 250 million preschool children are vitamin A deficient (WHO, 2013).
Vitamin A supplementation decreased NoV infection rates and clinical symptoms (Long et al., 2007). Moreover, the responses of various intestinal cytokines, including IL-6, IL-8, and IFN-α, were modified by vitamin A supplementation during NoV infection (Long et al., 2011). Chemokine (C–C motif) ligand 2 (CCL2), referred to as monocyte chemotactic protein 1 (MCP-1), was found to be upregulated in NoV-infected children but downregulated in vitamin A-supplemented children (Long et al., 2011). Anti-NoV effects of vitamin A via gut microbiota modulation have also been reported (Lee & Ko, 2016).
In this study, we aimed to determine transcriptome changes associated with vitamin A treatment during MNV infections, and to identify key host genes involved in vitamin A-mediated inhibition of NoV infections.
Materials and Methods
Cells, Viruses, and Reagents
The MNV strains murine norovirus-1 (MNV-1). CW1 and MNV-1.CR6 were obtained from Dr. Virgin's laboratory (School of Medicine, Washington University), and Huh-7-based NoV replicon-bearing cells (HG23 cells) were kindly provided by Dr. Chang (Chang et al., 2006). RAW 264.7 cells were maintained in Dulbecco’s minimal essential medium (DMEM) containing 10% fetal bovine serum (Gibco; Thermo Fisher Scientific), 10 mM HEPES, 10 mM sodium bicarbonate, 10 mM nonessential amino acids, and 50 μg/ml gentamicin. G418 (0.5 mg/ml) was also added to the medium for HG23 cells. Retinol (95144; Sigma-Aldrich) was applied to the cells after dilution with ethanol. Prior to in vitro experiments with retinol, cell viability was confirmed by PI Staining using the FACScan system (FACSCalibur; BD Biosciences).
Effect of Retinol and Simvastatin on MNV Replication in RAW 264.7 Cells
Retinol (100 U/ml) and simvastatin (5 µM) were applied to 0.01 MOI MNV-1.CW1-inoculated RAW 264.7 cells. MNV-1.CW1 replication was analyzed by plaque assay at 24 h after inoculation, as described previously (Lee et al., 2008).
Effects of MNV and Retinol on RNA Expression
The effects of retinol and MNV-1.CW1 treatment on RNA gene expression in RAW 264.7 cells were analyzed using microarrays. RAW 264.7 cells treated with retinol (50 U/ml) and MNV-1.CW1 (MOI 0.01) were incubated for 24 h in a 5% CO2 incubator. Total RNA was extracted using the easy-spin Total RNA Extraction Kit according to the manufacturer’s instructions. RNA purity and quantity were determined using a spectrophotometer (ND-1000; NanoDrop Technologies LLC). Whole genome expression microarray analysis with 4 × 44 K microarrays was performed by a commercial microarray analysis company (GenomicTree). RNA samples were amplified and labeled using the Low RNA Input Linear Amplification Kit PLUS (Agilent Technologies, Inc.), and then hybridized to whole mouse genome arrays using a Gene Expression Hybridization Kit (Agilent Technologies). Scanning and image analysis were performed using Microarray Scanner and Feature Extraction software (Agilent Technologies). Output files were imported into GeneSpring software (Agilent Technologies) for locally weighted scatterplot smoothing (LOWESS) normalization, clustering, and analysis of the expression data.
Confirmation of Gene Expression Levels
Expression levels were estimated using TaqMan qPCR for GBP1 and CCL6, and SYBR green qPCR for RIG-I. These genes were identified from the microarray results. The reaction mixture comprised the AgPath-ID One-Step RT-PCR kit (Ambion, Inc.). Amplification was performed using the 7300 Real-Time PCR System (Applied Biosystems). For amplification of the GBP1 and CCL6 genes, primers and probes were designed and synthesized manually: GBP1 (NM_010259.2): forward primer: 5-ATGCTCCAGCCTACTGTGTTTCC-3; reverse primer: 5-GAGCCTCAGAGGAATAGAACGAAAT-3; and probe: 5-FAM-CACTTTCTCCTTCTAAGAGTTTTAGAATGACTTCTACACAGAATGG-BHQ1-3; CCL6 (NM_009139.3): forward primer: 5-TTATTAGGGTAGTCTTTGGGGCTTTG-3; reverse primer: 5-ACAACTGGGAACCCACAAAGCT-3; and probe: 5-FAM-GTGTCTGGTTCTGATACAAGCTTAAGCCGGGT-BHQ1-3. The primer set for RIG-I, encoded by the Ddx58 gene, is shown in Table S1.
Animal Model
Twelve-week-old male ICR mice were purchased from KOATECH, and housed in an animal biosafety level 2 (ABL-2) facility at Seoul National University College of Medicine, South Korea. To exclude differences in the rate of metabolism of retinol, the active form of retinol, RA (R-2625; Sigma-Aldrich) was suspended in corn oil and administered orally to the mice (1 mg/kg/day) for 8 days. On day 7, the mice were infected perorally using 5 × 106 PFU of MNV-1.CW1 suspended in PBS by oral gavage. To confirm cytokine expression, ileum tissues were collected at 72 h after MNV-1.CW1 infection. Approximately 1 cm of terminal ileum with Peyer’s patches was stored in RNAlater® Solution (AM7021; Ambion) until RNA extraction.
Transcriptome Analysis
Prior to total RNA extraction, ileum samples were completely homogenized using a PT-2000 E homogenizer (PT-2000 E; Polytron Corp.). Total RNA was extracted using the easy-spin™ Total RNA Extraction Kit (iNtRON). cDNA was synthesized using the High Capacity RNA-to-cDNA Kit (Applied Biosystems) according to the manufacturer’s instructions. To estimate cytokine mRNA expression levels, the QuantiTect® SYBR® Green PCR Kit (204143; Qiagen) and 7300 Real Time PCR System (Applied Biosystems) were used. Analyzed cytokines and oligonucleotide primer sets are described in Table S1. The reaction mixture (25 µl) for real-time PCR was composed of 2 × QuantiTect SYBR Green PCR Master Mix (12.5 µl), primers (forward and reverse; both 50 pmol in 0.2 µl), RNase-free water (11.1 µl), and template DNA (1 µl). GAPDH was used as the internal control.
MNV Replication with Gene Knockdown by siRNA Treatment
To knock down the genes of interest, siRNAs for GBP1 (FlexiTube GeneSolution GS14468; Qiagen) and CCL6 (FlexiTube GeneSolution GS20305; Qiagen) were purchased, and siRNA for RIG-I was commercially synthesized (Bioneer Corp.) (Nasirudeen et al., 2011). AccuTarget™ Negative Control siRNA (SN-1001; Bioneer) was used as a negative control. RAW 264.7 cells (1 × 106 cells/well) were seeded onto 24-well plates and incubated for 6 h. siRNA (250 ng) was transfected using HiPerFect Transfection Reagent (301705; Qiagen) according to the manufacturer’s instructions. At 24 h post-transfection, MNV-1.CW1 (MOI 0.01) and retinol (20 U/ml) were applied. Replicated MNV-1.CW1 quantity was analyzed by plaque assay at 24 h post-treatment.
In vitro Test for NoV Inhibition by Retinol and CCL6
Huh-7-based NoV replicon-bearing cells (HG23 cells) were used to identify the effects of CCL6 on NoV replication. Recombinant murine CCL6 (1, 10, and 100 ng, 250–06; Peprotech) was applied to the HG23 cells. NoV replicon replication was quantified 72 h after treatment. For MNV replication, CCL6 treatment was repeated twice: 12 h prior to and after MNV infection. Recombinant murine IFN-β (8234-MB; R&D Systems) was used to compare the inhibitory effects.
Relative Replication of MNV Strains with CCL6 Knockdown
MNV-1.CW1 and MNV-1.CR6 strains were used for this experiment. RAW 264.7 (1 × 105) cells were seeded onto a plate and incubated for 6 h until transfection. Diluted siRNA for CCL6 (5, 10, and 25 nM) (FlexiTube GeneSolution GS20305; Qiagen) and HiPerFect Transfection Reagent (301705; Qiagen) were used for the formation of transfection complexes and incubated for 10 min at room temperature. Then, the transfection complexes were added onto the cells for gene silencing. At 30 h post-transfection, the cells were infected with MNV-1.CW1 and MNV-1.CR6 strains (MOI 0.01). MNV was quantified by plaque assay at 24 h post-infection.
Statistical Analysis
All features are expressed as means and standard deviations. To quantify in vivo mRNA levels relative to an internal control (GAPDH), the 2−ΔΔCt relative quantification method (ΔΔCt = (Ct.Target − Ct.GAPDH)Group1 − (Ct.Target − Ct.GAPDH)Group2) was used. Statistical significance was assessed using one-way analysis of variance (ANOVA) followed by Duncan’s post hoc test. P values < 0.05 were considered significant. All statistical analyses were performed using SPSS Statistics software 12.0 (IBM Corp.).
Results
Effects of Retinol on NoV Replication In vitro
To confirm whether vitamin A treatment inhibited NoV replication, we tested the inhibitory effects of retinol on MNV-1.CW1 replication in murine RAW 264.7 cells. As simvastatin exerts a positive effect on MNV replication by impairing IFN-α expression, as reported previously (Jung et al., 2012), the inhibitory effects of retinol were confirmed in simvastatin-treated RAW 264.7 cells. Simvastatin increased MNV replication 9.7-fold compared to negative controls, but retinol treatment decreased it 0.64-fold (Fig. 1A).

Identification of cytokine and chemokine on anti-MNV effect by vitamin A. A Effects of simvastatin and retinol on MNV replication. Treatment with retinol offset the effect of simvastatin on MNV replication. Retinol (100 U/ml) and simvastatin (5 µM) were applied to MNV-1.CW1-inoculated RAW 264.7 cells. MNV replication was measured by plaque assay at 24 h after inoculation. B Effects of retinol and MNV on mRNA levels. The effects of retinol and MNV-1.CW1 treatment on RNA gene expression in RAW 264.7 cells were analyzed using microarrays. Output files were imported into GeneSpring software (Agilent Technologies) for locally weighted scatterplot smoothing (LOWESS) normalization, clustering, and analysis of the expression data. Hierarchically clustered dendrograms of in vitro gene expression experiments with RAW 264.7 cells. A total of 41,174 genes were analyzed using microarrays. Eleven genes were upregulated twe-fold by MNV inoculation alone compared to retinol treatment alone or MNV inoculation during retinol treatment. One sample from each group was analyzed with 4 × 44 K microarrays. Twenty genes showed the reverse expression pattern. The intensity indicates the fold change in expression. C Confirmation of the expression of genes of interest identified by microarray analysis in vitro. The relative expression levels of CCL6 and GBP1 compared to a negative control were measured by real-time PCR. The expression of CCL6 was significantly increased by retinol during MNV inoculation, but GBP1 was decreased compared to only MNV inoculation. D MNV replication in RAW 264.7 cells treated with siRNAs targeting CCL6 and GBP1. MNV replication at 24 h after MNV infection was quantified by a plaque assay. Relative MNV replication was calculated compared to a siRNA negative control. All experiments were repeated three times. Different superscript letters indicate significant differences (P < 0.05) according to Duncan’s post hoc test
Transcriptome Profiling
To identify which innate immune response genes are involved in the inhibition of NoV replication, the transcript levels of 41,174 genes following retinol treatment and/or MNV infection were comprehensively monitored in RAW 264.7 cells using microarrays (Agilent Technologies). The genes were largely clustered into two groups based on their expression patterns. The 20 genes in cluster 1 were upregulated by MNV infection, but downregulated by retinol, even with MNV infection, compared to the negative control (Fig. 1B, Table S2). This cluster included four genes (Rig-1, H28, Ifih1, and Gbp1) thought to be involved in immune responses; six genes (Klhl17, Iigp1, Megf9, Slc17a6, Slc39a4, and 1500005I02Rik) that were membrane or signal factors, two that were cytoplasmic or nuclear factors (Stat1 and Ifi44), and one gene that had replication, recombination, and repair functions (BC013672). The remaining genes had other functions (Table S2). Eleven genes showed reverse expression patterns in response to retinol and MNV treatment (Fig. 1B, Table S3). Among them, four genes (Mansc1, Bglap1, Col14a1, and Ccl6) were related to “signal/secreted/extracellular region responses”, two were glycoprotein or membrane factors (Ppap2b and Gpr160), and one was an alternative splicing or splice variant factor (Hecw1); the remaining genes had other functions (Table S3).
Based on the gene expression results, we selected genes representative of each cluster for verification of their gene expression patterns following retinol treatment and/or MNV infection. These genes included GBP1 (guanylate-binding protein 1) and CCL6. Real-time reverse transcription PCR (RT-PCR) showed that MNV infection significantly increased GBP1 expression (3.27-fold), whereas retinol treatment significantly decreased it (0.13-fold) compared to the negative control (Fig. 1C). In contrast, retinol treatment significantly increased CCL6 expression (4.75-fold), while MNV inoculation increased it during RA administration (6.40-fold) (Fig. 1C). Considering the role of CCL6 in the immune response in sepsis (Coelho et al., 2007; Núñez et al., 2010), we hypothesized that elevated CCL6 levels upon RA administration would inhibit MNV replication.
MNV Replication with In Vitro CCL6 Knockdown
To confirm the effects of CCL6 on MNV replication, MNV-1.CW1 replication was measured using real-time RT-PCR in RAW 264.7 cells treated with siRNA targeting the CCL6 gene (Fig. 1D). MNV replication was significantly increased in CCL6-knockdown cells, but not in GBP1-knockdown cells. This correlation between the CCL6 expression pattern and MNV replication suggests a role for this chemokine, whose expression is increased by retinol treatment in response to MNV infection.
Cytokine mRNA Levels in Response to RA and MNV In Vivo Mouse Model
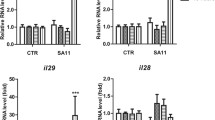
Based on our in vitro results using RAW 264.7 cells, innate immune response gene expression was investigated after MNV and RA inoculation in a mouse model. MNV inoculation without RA administration increased CCL6 levels (P = 0.016). RA administration significantly decreased RIG-I (P = 0.024), MDA-5 (P = 0.012), and IFN-β (P = 0.024) expression, and increased TNF-α (P = 0.006) and CCL6 (P = 0.024) expression (Fig. 2). MNV inoculation with RA administration significantly decreased the expression of RIG-I (P = 0.010), MDA-5 (P = 0.048), and CCL6 (P = 0.018) compared to MNV inoculation without RA (Fig. 2).

Effects of RA and MNV on cytokine and chemokine mRNA levels in vivo. RA was administered orally to the 12-week-old male ICR mice (1 mg/kg/day) for 8 days. On day 7, the mice were infected perorally using 5 × 106 PFU of MNV-1.CW1. To confirm cytokine expression, ileum tissues were collected at 72 h after MNV-1.CW1 infection. Ileum with Peyer’s patches was used to identify the mRNA expression level of mRNA. RA administration significantly increased the expression of TNF-α and CCL6, and decreased that of RIG-I, MDA-5, and IFN-β (RA: n = 7; NC: n = 5). MNV-1.CW1 inoculation plus RA administration significantly increased the expression of IFN-β, and decreased RIG-I and F4/80 (RA: n = 7; MNV + RA: n = 7). All experiments were repeated three times. Significance was analyzed by Mann–Whitney U-test. *P < 0.05
Inhibitory Effects of CCL6 on MNV Replication and HG23 Cell
To determine whether the retinol-induced increase in CCL6 has direct antiviral effects against NoV, NoV replication was monitored in RAW 264.7 and HG23 cells following CCL6 treatment. As there is no cell culture system to study human NoV, HG23 cells harboring a human NoV replicon were used to confirm the direct antiviral effect of CCL6. The application of 100 ng of CCL6 significantly decreased replication of the NoV genome in HG23 cells to 71.3% (Fig. 3B). However, direct inhibitory effects of CCL6 were not identified with MNV replication (Fig. 3A). Furthermore, IFN-β showed more significant anti-NoV effects in RAW 264.7 than HG23 cells (Fig. 3).

Antiviral effect of CCL6 and IFN-β against NoV replication. The direct antiviral effects of CCL6 and IFN-β were tested based on MNV replication in RAW 264.7 cells (A) and HG23 cells (B). Huh-7-based NoV replicon-bearing cells (HG23 cells) were used to identify the effects of CCL6 on NoV replication. Recombinant murine CCL6 was applied to the HG23 cells. NoV replicon replication was quantified 72 h after treatment. For MNV replication, CCL6 treatment was repeated twice: 12 h prior to and after MNV infection. Recombinant murine IFN-β was used to compare the inhibitory effects. CCL6 treatment significantly decreased the replication of HuNoV in HG23 cells but not MNV replication. All experiments were repeated three times. Significance was analyzed by Mann–Whitney U-test. *P < 0.05
MNV Strain Replication and CCL6 Knockdown
To uncover whether CCL6 plays an essential role in antiviral activity against NoV, we examined NoV replication in the presence of knocked down CCL6 expression. Prior to confirming replication of the MNV strain in RAW 264.7 cells, the relative expression of CCL6 was analyzed. The relative expression of CCL6 (5, 10, and 25 nM) was 0.56–0.66 compared to the negative control (Fig. 4A). The replication of MNV-1.CW1 was significantly increased 4.7–7.5-fold compared to the negative control (Fig. 4B). Furthermore, the increase of MNV-1.CR6 replication was significant only with 25 nM of CCL6 (Fig. 4B).

Relative replication of MNV strains with CCL6 knockdown in RAW 264.7 cells. Diluted siRNA targeting CCL6 with HiPerFect Transfection Reagent was transfected into RAW 264.7 cells, after which the cells were infected with strains MNV-1.CW1 and MNV-1.CR6 strains. MNV was quantified by a plaque assay at 24 h post-infection. A Confirmation of CCL6 knockdown in RAW 264.7 cells. The relative expression level of CCL6 (5, 10, and 25 nM) was 0.56–0.66 compared to the negative control, respectively. B Diluted siRNA targeting CCL6 (5, 10, and 25 nM) was transfected into cells that were then infected with strains MNV-1.CW1 and MNV-1.CR6 strains (MOI 0.01) at 30 h post-transfection. The quantification of MNV was analyzed by plaque assay at 24 h post-infection. All experiments were repeated three times. Significance was analyzed by Mann–Whitney U-test. *P < 0.05
Discussion
In this study, we demonstrated the inhibitory effects of vitamin A on NoV infection in vitro. We also demonstrated that innate immune response genes, including the gene encoding the chemokine CCL6, are likely related to the inhibitory effect of vitamin A on MNV multiplication. As no effective treatment or vaccine for NoV infection is currently available, the prevention of NoV infection through dietary supplementation has been considered important in public health. This study provides important knowledge regarding the role of vitamin A in preventing acute viral gastroenteritis.
Vitamin A supplementation has been suggested for NoV infection prevention, but the mechanism of the anti-NoV effect has not been identified (Long et al., 2007). Previous studies suggested that the antiviral effects of retinol in the gut result from acute-phase responses in innate immunity (Villamor & Fawzi, 2005). The secretion of key cytokines by macrophages, including TNF-α, IL-6, and IL-12, is also influenced by retinol (Arena et al., 1997; Long et al., 2011; Mathew & Sharma, 2000). In this study, TNF-α expression was significantly increased by RA. TNF-α is the primary mediator of inflammation, and also plays an important role in the induction of antiviral immune responses, such as CD8 + T cells (Tzeng et al., 2014). Moreover, TNF-α enhances the expression of specific chemokine gene, including CCL2, CCL5, CXC chemokine ligand (CXCL) 8, and CXCL10 (Veckman et al., 2006). Long et al. (2011) reported that specific chemokines, including TNF-α, CCL2, IL-6, and IL-8, were involved in the RA-induced immune response against NoV infections. Therefore, the antiviral immune response induced by TNF-α could be mediated by specific chemokines in NoV infections.
Chemokines play a fundamental role in the host defense mechanisms of the immune system, and their novel antiviral functions have been reported (Melchjorsen et al., 2003; Zlotnik & Yoshie, 2000). CCL2, CCL3, and CCL5 are almost always expressed in various virus infections (Melchjorsen et al., 2003). Interestingly, some chemokines are selectively expressed in different viral infections. For instance, CCL5 expression increases in respiratory syncytial virus infections, but it is not expressed in influenza virus infections (Dawson et al., 2000; Haeberle et al., 2001). CCL5 enhanced the antigen-specific Th1 CD4 + T cell-dependent immune response in the context of immunization with DNA vaccines encoding HSV-2 glyco-protein D (Sin et al., 2000). CXCL10 plays a key role in the immune response against central nervous system coronavirus infection through activation of natural killer cells (Trifilo et al., 2004).
Interestingly, CCL6 expression was significantly increased in retinol-treated mice in this study. Moreover, MNV replication was significantly increased in CCL6-knockdown cells. Although the mechanism of action of CCL6 in the immune response against NoV infection is not fully understood, it may involve inhibition of NoV replication. CCL6 and CCL9 were associated with retinoic X receptor alpha (RXRalpha) in macrophages, and played key roles in innate inflammatory responses (Núñez et al., 2010). Antiviral IFN responses are generally accompanied by the activation of a set of chemokine ligands (Melchjorsen et al., 2003). In particular, CCL6 expression was induced by type I and type II IFNs (Kanno et al., 2005; Nardi et al., 2009). Therefore, we suggest that CCL6 is the chemokine mediator of the immune response to NoV infection. However, this requires verification based on the mechanisms by which dietary vitamin A and simvastatin affect IFN and CCL6 levels in response to NoV multiplication.
In this study, the expression level of CCL6 was significantly increased after MNV infection in vivo in a mouse model, but not in vitro in MNV-inoculated RAW 264.7 cells. These contradictory results suggest the involvement of additional immune responses in the ileum against MNV infection that increased the CCL6 level after MNV infection in our mouse model. CCL6 is highly expressed in intestinal epithelial cells (Kotarsky et al., 2010). As gut microbiota modulation is involved in the inhibitory effects of vitamin A on NoV infection in mice (Lee & Ko, 2016, 2017), CCL6 activity may be related to the modulation of gut microbiota by vitamin A and NoV infection.
In previous studies, simvastatin, a cholesterol-lowering statin, had positive effects on NoV replication in the in vitro human norovirus (HuNoV) replicon system, and on viral shedding in an animal model (Chang, 2009; Jung et al., 2012). Although the mechanism remains unknown, impaired IFN-α expression by simvastatin increases MNV replication (Jung et al., 2012). In this study, no increase in NoV replication was observed with simvastatin treatment when retinol was administered. This led us to suspect that the antagonistic effect of vitamin A on NoV replication was much stronger than the agonistic effect of simvastatin, which could be related to the immune responses associated with IFNs. Meanwhile, the effect of vitamin A is epistatic to that of simvastatin in the same pathway affecting NoV replication. Vitamin A is important for the innate antiviral immunity mediated by type-I IFNs, including IFN-α and -β (Chen et al., 2012; Vlasova et al., 2013). Increased IFN-β expression was confirmed by retinol inoculation both in vivo and in vitro. Therefore, the regulation of IFN-β expression by vitamin A may be associated with NoV replication. This can account for the fact that the agonistic effects of simvastatin on NoV replication were not observed in the presence of retinol.
Direct antiviral effects of IFN have been identified in previous studies, for various viral infections (Samuel, 2001; Zorzitto et al., 2006). Chemokines such as CCL3, CCL4, and CCL5 showed significant direct antiviral effects against herpes simplex type 1 (Nakayama et al., 2006). In addition, CCL6 and CCL9 were induced by type-I IFNs (Nardi et al., 2009). In this study, CCL6 showed antiviral effects against HuNoV replication in HG23 cells, but did not affect MNV replication. Although we could not identify direct antiviral effects of CCL6, an indirect role through immunomodulation could be inferred.
The immunological functions of chemokines are typically performed through G protein-coupled chemokine receptors (Nibbs & Graham, 2013). CCL6, originally called macrophage inflammatory protein-related protein-1, has only been identified in rodents. Chemokines involved in human immune response to NoV infections have not been reported. In previous studies, activation of CCL6 via C–C chemokine receptor type 1 (CCR1) played a key role in the pathogenesis of IL-13-induced inflammation (Ma et al., 2004). The ligands of CCR1 are also known to regulate the activation of normal T expressed and secreted proteins (RANTES), monocyte chemoattractant protein 3, and myeloid progenitor inhibitory factor-1. The roles of chemokines in the human immune response to HuNoV, including the anti-NoV effects of CCL6, require further investigation.
In conclusion, the chemokine CCL6 may play a key role in the NoV-inhibiting effect of vitamin A treatment. Moreover, the antiviral effects of CCL6 might indirectly modulate the immune responses mediated by IFN-β. An understanding of the role of CCL6 in NoV infection could provide insights into the infectious etiology of NoV.
References
Arena, A., Capozza, A. B., Delfino, D., & Iannello, D. (1997). Production of TNF alpha and interleukin 6 by differentiated U937 cells infected with Leishmania major. The New Microbiologica, 20, 233–240.
Chang, K. O. (2009). Role of cholesterol pathways in norovirus replication. Journal of Virology, 83, 8587–8595.
Chang, K. O., Sosnovtsev, S. V., Belliot, G., King, A. D., & Green, K. Y. (2006). Stable expression of a Norwalk virus RNA replicon in a human hepatoma cell line. Virology, 353, 463–473.
Chen, S., Yang, Y., Yan, X., Chen, J., Yu, H., & Wang, W. (2012). Influence of vitamin A status on the antiviral immunity of children with hand, foot and mouth disease. Clinical Nutrition, 31, 543–548.
Coelho, A. L., Schaller, M. A., Benjamim, C. F., Orlofsky, A. Z., Hogaboam, C. M., & Kunkel, S. L. (2007). The chemokine CCL6 promotes innate immunity via immune cell activation and recruitment. The Journal of Immunology, 179, 5474–5482.
Dawson, T. C., Beck, M. A., Kuziel, W. A., Henderson, F., & Maeda, N. (2000). Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. The American Journal of Pathology, 156, 1951–1959.
Haeberle, H. A., Kuziel, W. A., Dieterich, H. J., Casola, A., Gatalica, Z., & Garofalo, R. P. (2001). Inducible expression of inflammatory chemokines in respiratory syncytial virus-infected mice: Role of MIP-1α in lung pathology. Journal of Virology, 75, 878–890.
Hall, J. A., Grainger, J. R., Spencer, S. P., & Belkaid, Y. (2011). The role of retinoic acid in tolerance and immunity. Immunity, 35, 13–22.
Jung, K., Wang, Q., Kim, Y., Scheuer, K., Zhang, Z., Shen, Q., Chang, K. O., & Saif, L. J. (2012). The effects of simvastatin or interferon-α on infectivity of human norovirus using a gnotobiotic pig model for the study of antivirals. PLoS ONE, 7, e41619.
Kanno, M., Suzuki, S., Fujiwara, T., Yokoyama, A., Sakamoto, A., Takahashi, H., Imai, Y., & Tanaka, J. (2005). Functional expression of CCL6 by rat microglia: A possible role of CCL6 in cell-cell communication. Journal of Neuroimmunology, 167, 72–80.
Karst, S. M., Wobus, C. E., Lay, M., Davidson, J., & Virgin, H. W. (2003). STAT1-dependent innate immunity to a Norwalk-like virus. Science, 299, 1575–1578.
Kotarsky, K., Sitnik, K. M., Stenstad, H., Kotarsky, H., Schmidtchen, A., Koslowski, M., Wehkamp, J., & Agace, W. W. (2010). A novel role for constitutively expressed epithelial-derived chemokines as antibacterial peptides in the intestinal mucosa. Mucosal Immunology, 3, 40–48.
Lee, H., & Ko, G. (2016). Antiviral effect of vitamin A on norovirus infection via modulation of the gut microbiome. Scientific Reports, 6, 25835.
Lee, H., & Ko, G. (2017). New perspectives regarding the antiviral effect of vitamin A on norovirus using modulation of gut microbiota. Gut Microbes, 8, 616–620.
Lee, J., Zoh, K., & Ko, G. (2008). Inactivation and UV disinfection of murine norovirus with TiO2 under various environmental conditions. Applied and Environmental Microbiology, 74, 2111–2117.
Lindesmith, L., Moe, C., Marionneau, S., Ruvoen, N., Jiang, X., Lindblad, L., Stewart, P., LePendu, J., & Baric, R. (2003). Human susceptibility and resistance to Norwalk virus infection. Nature Medicine, 9, 548–553.
Long, K. Z., Garcia, C., Ko, G., Santos, J. I., Al Mamun, A., Rosado, J. L., DuPont, H. L., & Nathakumar, N. (2011). Vitamin A modifies the intestinal chemokine and cytokine responses to norovirus infection in Mexican children. The Journal of Nutrition, 141, 957–963.
Long, K. Z., Garcia, C., Santos, J. I., Rosado, J. L., Hertzmark, E., Dupont, H. L., & Ko, G. (2007). Vitamin A supplementation has divergent effects on norovirus infections and clinical symptoms among mexican children. The Journal of Infectious Diseases, 196, 978–985.
Ma, B., Zhu, Z., Homer, R. J., Gerard, C., Strieter, R., & Elias, J. A. (2004). The C10/CCL6 chemokine and CCR1 play critical roles in the pathogenesis of IL-13-induced inflammation and remodeling. The Journal of Immunology, 172, 1872–1881.
Mathew, J. S., & Sharma, R. P. (2000). Effect of all-trans-retinoic acid on cytokine production in a murine macrophage cell line. International Journal of Immunopharmacology, 22, 693–706.
McCartney, S. A., Thackray, L. B., Gitlin, L., Gilfillan, S., Virgin, H. W., & Colonna, M. (2008). MDA-5 recognition of a murine norovirus. PLoS Pathogens, 4, e1000108.
Melchjorsen, J., Sørensen, L. N., & Paludan, S. R. (2003). Expression and function of chemokines during viral infections: From molecular mechanisms to in vivo function. Journal of Leukocyte Biology, 74, 331–343.
Mumphrey, S. M., Changotra, H., Moore, T. N., Heimann-Nichols, E. R., Wobus, C. E., Reilly, M. J., Moghadamfalahi, M., Shukla, D., & Karst, S. M. (2007). Murine norovirus 1 infection is associated with histopathological changes in immunocompetent hosts, but clinical disease is prevented by STAT1-dependent interferon responses. Journal of Virology, 81, 3251–3263.
Nakayama, T., Shirane, J., Hieshima, K., Shibano, M., Watanabe, M., Jin, Z., Nagakubo, D., Saito, T., Shimomura, Y., & Yoshie, O. (2006). Novel antiviral activity of chemokines. Virology, 350, 484–492.
Nardi, V., Naveiras, O., Azam, M., & Daley, G. Q. (2009). ICSBP-mediated immune protection against BCR-ABL-induced leukemia requires the CCL6 and CCL9 chemokines. Blood, 113, 3813–3820.
Nasirudeen, A. M. A., Wong, H. H., Thien, P., Xu, S., Lam, K. P., & Liu, D. X. (2011). RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Neglected Tropical Diseases, 5, e926.
Nibbs, R. J. B., & Graham, G. J. (2013). Immune regulation by atypical chemokine receptors. Nature Reviews Immunology, 13, 815–829.
Nice, T. J., Baldridge, M. T., McCune, B. T., Norman, J. M., Lazear, H. M., Artyomov, M., Diamond, M. S., & Virgin, H. W. (2015). Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science, 347, 269–273.
Núñez, V., Alameda, D., Rico, D., Mota, R., Gonzalo, P., Cedenilla, M., Fischer, T., Boscá, L., Glass, C. K., Arroyo, A. G., & Ricote, M. (2010). Retinoid X receptor α controls innate inflammatory responses through the up-regulation of chemokine expression. Proceedings of the National Academy of Sciences of the United States of America, 107, 10626–10631.
Patel, M. M., Widdowson, M. A., Glass, R. I., Akazawa, K., Vinjé, J., & Parashar, U. D. (2008). Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerging Infectious Diseases, 14, 1224–1231.
Samuel, C. E. (2001). Antiviral actions of interferons. Clinical Microbiology Reviews, 14, 778–809.
Sin, J., Kim, J. J., Pachuk, C., Satishchandran, C., & Weiner, D. B. (2000). DNA vaccines encoding interleukin-8 and RANTES enhance antigen-specific Th1-type CD4+ T-cell-mediated protective immunity against herpes simplex virus type 2 in vivo. Journal of Virology, 74, 11173–11180.
Takeuchi, O., & Akira, S. (2009). Innate immunity to virus infection. Immunological Reviews, 227, 75–86.
Thornton, K. A., Mora-Plazas, M., Marín, C., & Villamor, E. (2014). Vitamin A deficiency is associated with gastrointestinal and respiratory morbidity in school-age children. The Journal of Nutrition, 144, 496–503.
Trifilo, M. J., Montalto-Morrison, C., Stiles, L. N., Hurst, K. R., Hardison, J. L., Manning, J. E., Masters, P. S., & Lane, T. E. (2004). CXC chemokine ligand 10 controls viral infection in the central nervous system: Evidence for a role in innate immune response through recruitment and activation of natural killer cells. Journal of Virology, 78, 585–594.
Tzeng, H. T., Tsai, H. F., Chyuan, I. T., Liao, H. J., Chen, C. J., Chen, P. J., & Hsu, P. N. (2014). Tumor necrosis factor-alpha induced by hepatitis B virus core mediating the immune response for hepatitis B viral clearance in mice model. PLoS ONE, 9, e103008.
Veckman, V., Osterlund, P., Fagerlund, R., Melen, K., Matikainen, S., & Julkunen, I. (2006). TNF-α and IFN-α enhance influenza-A-virus-induced chemokine gene expression in human A549 lung epithelial cells. Virology, 345, 96–104.
Villamor, E., & Fawzi, W. W. (2005). Effects of vitamin A supplementation on immune responses and correlation with clinical outcomes. Clinical Microbiology Reviews, 18, 446–464.
Vimont, A., Fliss, I., & Jean, J. (2015). Efficacy and mechanisms of murine norovirus inhibition by pulsed-light technology. Applied and Environmental Microbiology, 81, 2950–2957.
Vinje, J., Estes, M. K., Esteves, P., Green, K. Y., Katayama, K., Knowles, N. J., L’Homme, Y., Martella, V., Vennema, H., White, P. A., et al. (2019). ICTV virus taxonomy profile: Caliciviridae. Journal of General Virology, 100, 1469–1470.
Vlasova, A. N., Chattha, K. S., Kandasamy, S., Siegismund, C. S., & Saif, L. J. (2013). Prenatally acquired vitamin A deficiency alters innate immune responses to human rotavirus in a gnotobiotic pig model. The Journal of Immunology, 190, 4742–4753.
WHO World Health Organization. (2013). Vitamin A deficiency, micronutrient deficiencies. Geneva: World Health Organization.
Zlotnik, A., & Yoshie, O. (2000). Chemokines: A new classification system and their role in immunity. Immunity, 12, 121–127.
Zorzitto, J., Galligan, C. L., Ueng, J. J. M., & Fish, E. N. (2006). Characterization of the antiviral effects of interferon-α against a SARS-like coronoavirus infection in vitro. Cell Research, 16, 220–229.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant (No. 2011-0000177) funded by the Korean government (MEST), and the Korea Initiative for Fostering University of Research and Innovation Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (No. NRF-2020M3H1A1073304).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interest to report.
Ethical approval
All experimental protocols were approved by the Seoul National University Institutional Animal Care and Use Committee (SNU-111208-4).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lee, H., Lee, G., Cho, YH. et al. Chemokine CCL6 Plays Key Role in the Inhibitory Effect of Vitamin A on Norovirus Infection. J Microbiol. 61, 579–587 (2023). https://doi.org/10.1007/s12275-023-00047-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12275-023-00047-3