
Abstract
Nineteen pyrrolo[1,2-h][1,7]naphthyridinones and pyrido[2,3-c]pyrrolo[1,2-a]azepinones were synthesized as new tricyclic systems in which the pyridine ring is annelated to the 6,7-dihydroindolizin-8(5H)-one and 5,6,7,8-tetrahydro-9H-pyrrole[1,2-a]azepine-9-one moieties to obtain potential photosensitizing agents. They were tested for their photoantiproliferative activity on a triple-negative breast cancer cell line, MDA-MB-231, in the dark and under UVA light (2.0 J/cm2). We demonstrated that their toxicity, only when exposed to light, was primarily due to the generation of reactive oxygen species while their photodegradation products were not responsible for their activity. The most active compounds exhibited photocytotoxicity with IC50 values at low micromolar level inducing a decrease in the intracellular content of thiol, thus triggering cancer cell death through apoptosis. All the pyridone derivatives revealed to be pure photosensitizers with preferential photocytotoxic activity towards cancerous over healthy cells. Altogether, the results obtained confirm pyrrolo[1,2-h][1,7]naphthyridinones and pyrido[2,3-c]pyrrolo[1,2-a]azepinones as promising photosensitisers against triple-negative breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer represents one of the most challenging tasks in drug discovery, and despite several progress have been done, many efforts are still needed. Among the different kind of applications in the treatment of cancer and benign tumours, the use of light is considered a promising non-invasive modality with high selectivity towards malignant target versus normal cells. It can be selectively applied onto a region by illuminating the specific lesion using light with a proper wavelength to activate a tumour-targeted photosensitizer (PS). The latter, by in situ generation of highly cytotoxic reactive oxygen species (ROS), will be able to selectively destroy tumour tissue offering much lower toxic side effects. Over the years there has been a wide number of studies investigating different types of PSs, but the only approved one for clinical use have a tetrapyrrole backbone, such as porphyrins and their analogues. However, their large size is often related to some challenges such as limited tumour specificity and in some cases very important photosensitivity reactions caused by PS accumulation in off-target tissues. Small molecules show several advantages as they typically possess fast distribution throughout the body, rapid clearance, and target accumulation at the tumour site. During our studies aiming at the development of new bioactive heterocyclic compounds (Spanò et al. 2021a, b; Barreca et al. 2022b; Cilibrasi et al. 2022; Labbozzetta et al. 2022), we explored several classes of PSs, and some of them showed high potency, with outstanding selectivity index versus tumour cells inducing mitochondrial apoptotic death (Spanò et al. 2017). We recently reported our studies on two classes of small heterocyclic compounds, namely pyrimido[5,4-g]indolizines 1 and pyrimido[4,5-c]pyrrolo[1,2-a]azepines 2 (Fig. 1) and their antiproliferative activity against the highly refractory triple negative human breast cancer (MDA-MB-231) and vesical human tumour (T24) cell lines, in the dark and under UVA light irradiation. Results indicated very promising antitumour effect, with IC50 values up to nanomolar level and strong induction of reactive oxygen species (ROS) and a thiol redox stress responsible of apoptotic cell death (Barreca et al. 2022a). However, some compounds belonging to these series showed activity already in the dark, which is considered not optimal for their use as PSs. In order to extend our insight around their chemical space and inspired by our previous studies indicating the pyridine-2-one moiety as a valuable structural feature within our photoactivable compounds (Barraja et al. 2006, 2010, 2011; Spanò et al. 2017), we decided to explore the effect of the replacement of the pyrimidine moiety generating new classes of tricyclic compounds containing the indolizine and azepine structures incorporating the pyridine-2-one moiety. The choice was supported by our previous evidences related to pyrroloquinolinones 3 and their positional isomers 4, 5 (Fig. 1) which were able to produce phototoxicity in the submicromolar–micromolar range (IC50 0.4–17.5 μM, 0.2–17.5 μM and 0.5–9.3 μM, respectively), high ROS level with the involvement of mitochondria and lysosomes (Barraja et al. 2006, 2010, 2011; Spanò et al. 2017) without generating DNA strand breaks and DNA oxidative damages which represents a great disadvantage in this kind of applications. The enlargement of the central ring of the latter class of derivatives led to pyrrolo[3’,2’:6,7]cyclohepta[1,2-b]pyridines 6 (Fig. 1), which maintained phototoxic activity at submicromolar–micromolar level (EC50 0.25–10.70 μM). It was demonstrated that they were able to trigger a rapid apoptotic process through the induction of ROS production with a clear involvement of mitochondrial disfunction and lysosomes.

Previously synthetized pyrrolo pyrimido (1, 2) and pyrrolo pyridine-2-one systems (3–6), and new pyridine scaffolds: pyrrolo[1,2-h][1,7]naphthyridinones (7) and pyrido[2,3-c]pyrrolo[1,2-a]azepinones (8)
In this paper we wish to report the synthesis and the evaluation of the photoantiproliferative activity of the new tricyclic systems pyrrolo[1,2-h][1,7]naphthyridinones 7 and pyrido[2,3-c]pyrrolo[1,2-a]azepinones 8 (Fig. 1) and their evaluation as PSs against a very difficult type of breast cancer (BC) to be treated, the triple-negative breast cancer (TNBC).
BC can be described as an etiologically and clinically heterogeneous group of tumours originating from mammary epithelial cells (Lüönd et al. 2021). They can be divided into five molecular subtypes: luminal A, luminal B, HER2-enriched, basal-like (also known as triple-negative breast cancer) and normal-like breast cancer (Ali et al. 2020). These subtypes show differences in incidence, age at diagnosis, prognosis, therapeutic response, and disease progression (Liu et al. 2018). TNBC is an invasive breast carcinoma and represents around 15–20% of newly diagnosed breast cancer (Criscitiello et al. 2012). TNBC is associated with young age at diagnosis (< 40 years) and early metastases despite optimal adjuvant treatment. The metastatic behaviour is predominant in the lungs, central nervous system, and liver (Bergin and Loi 2019). Significant heterogeneity exists within the TNBC class, contrary to the basal-like subtype cancers that present a similar gene expression (Yao et al. 2017). Despite differences within this group, all TNBC subtypes are characterized by the absence of oestrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) expression (Sporikova et al. 2018). Therefore, among all BC subtypes, TNBC has the most limited therapeutic options (Tong et al. 2018) as patients with TNBC do not benefit from therapies that are designed to target the above biomarkers (Tong et al. 2018). All the new synthesized derivatives were tested on TNBC cells, namely MDA-MB-231 human cancer cells, in the dark and upon UVA light. Furthermore, in order to assess their selectivity against tumour cells, all derivatives were additionally screened on a non-transformed human cell line (HEK293 cells). Finally, selected mechanistic investigations were also performed with the aim to deeply characterize their mechanism of action and to evaluate the cell death pathway triggered by the most representative compounds.
Materials and methods
Chemistry
All melting points were taken on a Büchi melting point M-560 apparatus. IR spectra were determined in bromoform with a Shimadzu FT/IR 8400S spectrophotometer. 1H and 13C NMR spectra were measured at 200 and 50.0 MHz, respectively, in DMSO-d6 or CDCl3 solution using a Bruker Avance II series 200 MHz spectrometer. Column chromatography was performed with Merck silica gel (230 − 400 mesh ASTM) or a Büchi Sepacor chromatography module (prepacked cartridge system). Elemental analyses (C, H, N) were within ± 0.4% of theoretical values and were performed with a VARIO EL III elemental analyzer. The purity of all the tested compounds was > 95%, determined by HPLC (Agilent 1100 series).
Compounds 9a–e were prepared according to published procedure (Barreca et al. 2022a).
General procedure for the synthesis of pyrrolo[1,2-h][1,7]naphthyridinones (7a–c) and pyrido[2,3-c]pyrrolo[1,2-a]azepinones (8a, b)
To a solution of enamiketones 9a–e (2.76 mmol) in anhydrous ethanol (20 mL) phenylsulfonylacetonitrile (Sigma-Aldrich, Schnelldorf, Germany) (0.75 g, 4.14 mmol) was added under nitrogen atmosphere. The reaction mixture was heated under reflux for 24 h. After cooling, the solvent was removed under reduced pressure. The crude product was recrystallized from diethyl ether.
3-(benzenesulfonyl)-5,6-dihydropyrrolo[1,2-h][1,7]naphthyridin-2(1H)-one (7a)
This compound was obtained by reaction of 9a. Yellow solid; yield: 64%; mp: 317–318 °C; IR cm−1: 3119 (NH), 1641 (CO); 1H NMR (DMSO-d6, 200 MHz, ppm): δ 2.95 (t, 2H, J = 6.7 Hz, CH2), 4.11 (t, 2H, J = 6.7 Hz, CH2) 6.22–6.25 (m, 1H, Ar), 7.13–7.16 (m, 2H, Ar), 7.54–7.71 (m, 3H, H-Ar), 7.95–8.00 (m, 2H, Ar), 8.20 (s, 1H, H-4), 12.38 (s, 1H, NH). 13C NMR (DMSO-d6, 50 MHz, ppm): δ 25.4, 43.4, 106.2, 110.1, 111.7, 121.7, 127.0, 127.8, 128.7, 133.0, 135.5, 140.8, 141.9, 143.7, 157.2. Anal. Calcd. for C17H14N2O3S: C, 62.56; H, 4.32; N, 8.58. Found: C, 62.72; H, 4.24; N, 8.41.
Ethyl 3-(benzenesulfonyl)-2-oxo-1,2,5,6-tetrahydropyrrolo[1,2-h][1,7]naphthyridine-8-carboxylate (7b)
This compound was obtained by reaction of 9b. Brown solid; yield: 70%; mp: 145–146 °C; IR cm−1: 3365 (NH), 1718 (CO), 1646 (CO); 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.29 (t, 3H, J = 7.1 Hz, CH3), 3.01 (t, 2H, J = 7.0 Hz, CH2), 4.27 (q, 2H, J = 7.1 Hz, CH2), 4.54 (t, 2H, J = 7.0 Hz, CH2), 6.65 (d, 1H, J = 4.0 Hz, Ar), 7.16 (d, 1H, J = 4.0 Hz, Ar), 7.55–7.73 (m, 3H, Ar), 7.96–8.01 (m, 2H, Ar), 8.29 (s, 1H, H-4), 12.58 (s, 1H, NH); 13C NMR (DMSO-d6, 50 MHz, ppm): δ 14.2, 24.8, 42.1, 60.3, 108.4, 110.4, 117.6, 118.7, 120.1, 125.5, 128.0, 128.8, 133.3, 138.9, 140.3, 144.1, 157.1, 159.9. Anal. Calcd. for C20H18N2O5S: C, 60.29; H, 4.55; N, 7.03. Found: C, 60.17; H, 4.69; N, 6.85.
Propan-2-yl 3-(benzenesulfonyl)-2-oxo-1,2,5,6-tetrahydropyrrolo[1,2-h][1,7]naphthyridine-8-carboxylate (7c)
This compound was obtained by reaction of 9c. White solid; yield: 71%, mp: 332–333 °C; IR: 3125 (NH), 1703 (CO), 1652 (CO) cm−1; 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.29 (6H, d, J = 6.2 Hz, 2 × CH3), 3.00 (2H, t, J = 6.7 Hz, CH2), 4.54 (2H, t, J = 6.7 Hz, CH2), 5.07–5.13 (1H, m, CH), 6.91 (1H, d, J = 4.1 Hz, Ar), 7.14 (1H, d, J = 4.1 Hz, Ar), 7.56–7.69 (3H, m, Ar), 7.99 (2H, d, J = 7.5 Hz, Ar), 8.29 (1H, s, H-4), 12.57 (1H, s, NH); 13C NMR (DMSO-d6, 50 MHz, ppm): δ 22.1, 25.3, 42.6, 68.3, 107.5, 110.8, 118.0, 123.9, 126.3, 128.5, 129.2, 133.8 (2 × C), 140.8, 145.8, 149.7, 157.5, 160.0. Anal. Calcd. for C21H20N2O5S: C, 61.15; H, 4.89; N, 6.79. Found: C, 61.33; H, 5.02; N, 6.64.
Ethyl 3-(benzenesulfonyl)-2-oxo-1,5,6,7-tetrahydro-2H-pyrido[2,3-c]pyrrolo[1,2-a]azepine-9-carboxylate (8a)
This compound was obtained by reaction of 9d. Yellow solid; yield: 61%, mp: 340–341 °C; IR: 3131 (NH), 1703 (CO), 1652 (CO) cm−1; 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.29 (3H, t, J = 7.1 Hz, CH3), 2.23–2.26 (2H, m, CH2), 2.32–2.59 (2H, s, CH2), 4.27 (2H, q, J = 7.1 Hz, CH3), 4.38 (2H, t, J = 6.1 Hz, CH2), 6.64 (1H, d, J = 4.2 Hz, Ar), 6.94 (1H, d, J = 4.2 Hz, Ar), 7.57–7.70 (3H, m, Ar), 8.00 (2H, d, J = 6.9 Hz, Ar), 8.33 (1H, s, H-4), 12.54 (1H, s, NH); 13C NMR (DMSO-d6, 50 MHz, ppm): δ 14.7, 27.2, 31.8, 44.0, 60.6, 112.7, 117.0, 117.7, 125.7, 126.5, 128.7, 129.3, 133.3, 133.8, 140.7, 145.2, 146.0, 157.6, 160.5. Anal. Calcd. for C21H20N2O5S: C, 61.15; H, 4.89; N, 6.79. Found: C, 61.02; H, 4.69; N, 6.91.
Propan-2-yl 3-(benzenesulfonyl)-2-oxo-1,5,6,7-tetrahydro-2H-pyrido[2,3-c]pyrrolo[1,2-a]azepine-9-carboxylate (8b)
This compound was obtained by reaction of 9e. White solid; yield: 54%, mp: 237–238 °C; IR: 3131 (NH), 1697 (CO), 1646 (CO) cm−1; 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.30 (6H, d, J = 6.1 Hz, 2 × CH3), 2.12–2.34 (2H, m, CH2), 2.42–2.51 (2H, m, CH2), 4.29–4.43 (2H, m, CH2), 5.04–5.16 (1H, m, CH), 6.63 (1H, d, J = 4.1 Hz, Ar), 6.91 (1H, d, J = 4.1 Hz, Ar), 7.57–7.73 (3H, m, Ar), 8.01 (2H, d, J = 6.9 Hz, Ar), 8.33 (1H, s, H-4), 12.53 (1H, s, NH); 13C NMR (DMSO-d6, 50 MHz, ppm): δ 21.7, 26.7, 31.3, 43.6, 67.6, 102.8, 112.1, 116.4, 121.4, 125.6, 128.2, 128.8, 133.4, 137.0, 140.1, 145.5, 155.7, 157.2, 159.7. Anal. Calcd. for C22H22N2O5S: C, 61.96; H, 5.20; N, 6.57. Found: C, 62.22; H, 5.05; N, 6.68.
General procedure for the synthesis of N-methyl derivatives (7d–f and 8c, d) and O-methyl derivatives (7g–i and 8e, f)
To a solution of derivatives 7a–c and 8a, b (15 mmol) in anhydrous DMF (20 mL), NaH (Sigma-Aldrich, Schnelldorf, Germany) (0.64 g, 16 mmoli) was added at 0 °C and the reaction mixture was stirred at room temperature for 6 h. Then, iodomethane (Sigma-Aldrich, Schnelldorf, Germany) (16 mmol) was added at 0 °C and the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was poured onto crushed ice. The precipitate was filtered off and dried, in absence the solution was extracted with ethyl acetate (3 × 30 mL). The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. The crude product, containing N-methyl substituted derivatives 7d–f and 8c, d and O-methyl 7g–i and 8e, f, was purified by column chromatography (DCM/AcOEt 95:5).
1-Methyl-3-(benzenesulfonyl)-5,6-dihydropyrrolo[1,2-h][1,7]naphthyridin-2(1H)-one (7d)
This compound was obtained by reaction of 7a. Yellow solid; yield: 57%, mp: 268–269 °C; IR: 1658 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 3.11 (2H, t, J = 6.7 Hz, CH2), 3.95 (3H, s, CH3), 4.14 (2H, t, J = 6.7 Hz, CH2), 6.31–6.35 (1H, m, Ar), 6.81–6.91 (1H, m, Ar), 6.92–6.95 (1H, m, Ar), 7.46–7.58 (3H, m, Ar), 8.11–8.16 (2H, m, Ar), 8.21 (1H, s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ 29.7, 34.6, 44.6, 108.2, 110.0, 115.7, 122.0, 122.5, 126.1, 128.6, 128.8, 133.0, 140.3, 141.5, 144.5, 157.8. Anal. Calcd. for C18H16N2O3S: C, 63.51; H, 4.74; N, 8.23. Found: C, 63.62; H, 4.55; N, 8.07.
Ethyl 3-(benzenesulfonyl)-1-methyl-2-oxo-1,2,5,6-tetrahydropyrrolo[1,2-h][1,7]naphthyridine-8-carboxylate (7e)
This compound was obtained by reaction of 7b. Yellow solid; yield: 53%, mp: 250–251 °C; IR: 1709 (CO), 1658 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.38 (3H, t, J = 7.1 Hz, CH3), 2.93 (2H, t, J = 6.4 Hz, CH2), 3.77 (3H, s, CH3), 4.35 (2H, q, J = 7.1 Hz, CH2), 4.68 (2H, t, J = 6.4 Hz, CH2), 6.76 (1H, d, J = 4.4 Hz, Ar), 7.03 (1H, d, J = 4.4 Hz, Ar), 7.47–7.64 (3H, m, 3H-Ar), 8.12–8.18 (2H, m, 2H-Ar), 8.27 (1H, s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ 14.4, 27.7, 35.2, 41.8, 60.9, 110.7, 113.9, 117.3, 125.4, 125.5, 127.1, 128.6, 129.0, 133.3, 139.9, 141.5, 143.1, 157. 8, 160.5. Anal. Calcd. for C21H20N2O5S: C, 61.15; H, 4.89; N, 6.79. Found: C, 60.95; H, 5.02; N, 6.66.
Propan-2-yl 3-(benzenesulfonyl)-1-methyl-2-oxo-1,2,5,6-tetrahydropyrrolo[1,2-h][1,7]naphthyridine-8-carboxylate (7f)
This compound was obtained by reaction of 7c. Yellow solid; yield: 55%, mp: 207–208 °C; IR: 1703 (CO), 1658 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.36 (6H, d, J = 6.2 Hz, 2 × CH3), 2.92 (2H, t, J = 7.2 Hz, CH2), 3.77 (3H, s, CH3), 4.68 (2H, t, J = 7.2 Hz, CH2), 5.18–5.24 (1H, m, CH), 6.75 (1H, d, J = 4.4 Hz, Ar), 7.01 (1H, d, J = 4.4 Hz, Ar), 7.47–7.63 (3H, m, Ar), 8.11–8.17 (2H, m, Ar), 8.27 (1H,s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ 21.9, 27.7, 35.1, 41.8, 68.5, 110.7, 113.8, 117.2, 125.4, 125.), 126.9, 128.6, 129.1, 133.3, 139.9, 141.5, 143.2, 157.8, 160.1. Anal. Calcd. for C22H22N2O5S: C, 61.96; H, 5.20; N, 6.57. Found: C, 62.09; H, 5.34; N, 6.29.
2-methoxy-3-(phenylsulfonyl)-5,6-dihydropyrrolo[1,2-h][1,7]naphthyridine (7g)
This compound was obtained by reaction of 7a. White solid; yield: 37%; mp: 210–211 °C; 1H NMR (CDCl3, 200 MHz, ppm): δ 3.11 (2H, t, J = 6.6 Hz, CH2), 3.95 (3H, s, CH3), 4.12 (2H, t, J = 6.6 Hz, CH2), 6.25–6.28 (1H, m, Ar), 6.81 (1H, s, Ar), 6.88–6.90 (1H, m, Ar), 7.44- 7.61 (3H, m, Ar), 7.97–8.01 (2H, m, Ar), 8.15 (1H, s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ 27.4, 43.9, 53.9, 108.2, 110.0, 110.3, 117.5, 118.8, 124.4, 128.4, 128.6, 129.3, 133.0, 138.3, 141.1, 150.4, 159.2. Anal. Calcd. for C18H16N2O3S: C, 63.51; H, 4.74; N, 8.23. Found: C, 63.43; H, 4.88; N, 7.99.
Ethyl 3-(benzenesulfonyl)-2-methoxy-5,6-dihydropyrrolo[1,2-h][1,7]naphthyridine-8-carboxylate (7h)
This compound was obtained by reaction of 7b. Light yellow solid; yield: 35%, mp: 157–158 °C; IR: 1697 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.37 (3H, t, J = 7.1 Hz, CH3), 3.13 (2H, t, J = 6.9 Hz, CH2), 3.96 (3H, s, CH3), 4.32 (2H, q, J = 7.1 Hz, CH2), 4.67 (2H, t, J = 6.9 Hz, CH2), 6.88 (1H, d, J = 4.1 Hz, Ar), 7.01 (1H, d, J = 4.1 Hz, Ar), 7.46–7.64 (3H, m, 3H-Ar), 7.98–8.03 (2H, m, Ar), 8.23 (1H, s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ 14.4, 26.9, 42.2, 54.0, 60.4, 109.5, 118.4, 119.2, 121.0, 124.9, 128.5, 128.7, 133.3, 134.6, 138.5, 140.6, 149.2, 159.11, 161.1. Anal. Calcd. for C21H20N2O5S: C, 61.15; H, 4.89; N, 6.79. Found: C, 61.31; H, 4.76; N, 6.90.
Propan-2-yl 3-(benzenesulfonyl)-2-methoxy-5,6-dihydropyrrolo[1,2-h][1,7]naphthyridine-8-carboxylate (7i)
This compound was obtained by reaction of 7c. Yellow solid; yield: 44%, mp: 168–169 °C; IR: 1692 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.35 (6H, d, J = 6.2 Hz, 2 × CH3), 3.31 (2H, t, J = 6.9 Hz, CH2), 3.97 (3H, s, CH3), 4.67 (2H, t, J = 6.9 Hz, CH2), 5.13–5.26 (1H, m, CH), 6.88 (1H, d, J = 4.1 Hz Ar), 6.99 (1H, d, J = 4.1 Hz, Ar), 7.47–7.63 (3H, m, Ar), 8.03 (2H, d, J = 6.6 Hz Ar), 8.22 (1H,s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.0, 26.9, 42.2, 54.0, 67.9, 109.4, 118.2, 119.1., 120.9, 125.4, 128.5, 128.7, 133.3, 134.5, 138.5, 140.7, 149.2, 159.1, 160.7. Anal. Calcd. for C22H22N2O5S: C, 61.96; H, 5.20; N, 6.57. Found: C, 61.82; H, 5.41; N, 6.28.
Ethyl 3-(benzenesulfonyl)-1-methyl-2-oxo-1,5,6,7-tetrahydro-2H-pyrido[2,3-c]pyrrolo[1,2-a]azepine-9-carboxylate (8c)
This compound was obtained by reaction of 8a. Yellow solid; yield: 55%, mp: 238–239 °C; IR: 1697 (CO), 1658 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.39 (3H, t, J = 7.1 Hz, CH3), 2.05–2.19 (2H, m, CH2), 2.37–2.62 (2H, m, CH2), 3.60 (3H, s, CH3), 4.34(2H, q, J = 7.1 Hz, CH2), 5.35–5.45 (2H, m, CH2), 6.37 (1H, d, J = 4.2 Hz, Ar), 7.04 (1H, d, J = 4.2 Hz, Ar), 7.49–7.65 (3H, m, Ar), 8.11–8.18 (2H, m, Ar), 8.28 (1H, s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ 14.4, 28.3, 31.9, 35.3, 43.3, 60.6, 112.6, 116.3, 116.8, 124.4, 127.6, 128.6, 129.1, 130.6, 133.4, 139.7, 143.3, 145.8, 157.6, 160.7. Anal. Calcd. for C22H22N2O5S: C, 61.96; H, 5.20; N, 6.57. Found: C, 62.11; H, 5.07; N, 6.69.
Propan-2-yl 3-(benzenesulfonyl)-1-methyl-2-oxo-1,5,6,7-tetrahydro-2H-pyrido[2,3-c]pyrrolo[1,2-a]azepine-9-carboxylate (8d)
This compound was obtained by reaction of 8b. Yellow solid; yield: 51%, mp: 253–254 °C; IR: 1697 (CO), 1658 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.37 (6H, d, J = 6.2 Hz, 2 × CH3), 2.08–2.20 (2H, m, CH2), 2.41–2.61 (2H, m, CH2), 3.60 (3H, s, CH3), 5.15–5.44 (3H, m, CH2 and CH), 6.35 (1H, d, J = 4.1 Hz, Ar), 7.02 (1H, d, J = 4.1 Hz, Ar), 7.54–7.62 (3H, m, Ar), 8.14–8.19 (2H, m, Ar), 8.29 (1H,s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.0, 28.4, 31.8, 35.3, 43.3, 68.2, 112.5, 116.3, 117.8, 124.9, 127.7, 128.6, 129.2, 130.5, 133.4, 139.7, 143.3, 145.9, 157.6, 160.2. Anal. Calcd. for C23H24N2O5S: C, 62.71; H, 5.49; N, 6.36. Found: C, 62.57; H, 5.22; N, 6.58.
Ethyl 3-(benzenesulfonyl)-2-methoxy-6,7-dihydro-5H-pyrido[2,3-c]pyrrolo[1,2-a]azepine-9-carboxylate (8e)
This compound was obtained by reaction of 8a. White solid; yield: 43%, mp: 157–158 °C; IR: 1697 cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.38 (3H, t, J = 7.1 Hz, CH3), 2.31–2.45 (2H, m, CH2), 2.70 (2H, t, J = 7.0 Hz, CH2), 3.96 (3H, s, CH3), 4.27–4.45 (4H, m, 2 × CH2), 6.62 (1H, d, J = 4.1 Hz, Ar), 7.01 (1H, d, J = 4.1 Hz, Ar), 7.49–7.65 (3H, m, Ar), 8.01–8.06 (2H, m, Ar), 8.27 (1H,s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ δ 14.4, 28.7, 31.5, 43.8, 51.1, 60.3, 111.0, 117.4, 121.6, 125.0, 126.5, 128.6, 128.7, 133.4, 140.0, 140.5, 140.6, 153.4, 158.4, 161.1. Anal. Calcd. for C22H22N2O5S: C, 61.96; H, 5.20; N, 6.57. Found: C, 61.83; H, 5.01; N, 6.75.
Propan-2-yl 3-(benzenesulfonyl)-2-methoxy-6,7-dihydro-5H-pyrido[2,3-c]pyrrolo[1,2-a]azepine-9-carboxylate (8f)
This compound was obtained by reaction of 8b. White solid; yield: 46%, mp: 211–212 °C; IR: 1696 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.35 (6H, d, J = 6.2 Hz, 2 × CH3), 2.35–2.44 (2H, m, CH2), 2.64–2.73 (2H, m, CH2), 3.95 (3H, s, CH3), 4.41 (2H, t, J = 6.5 Hz, CH2), 5.13–5.26 (1H, m, CH), 6.62 (1H, d, J = 4.1 Hz, Ar), 7.00 (1H, d, J = 4.1 Hz, Ar), 7.49–7.65 (3H, m, Ar), 8.0 (2H, d, J = 6.8 Hz, Ar), 8.27 (1H,s, H-4); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.1, 28.7, 31.5, 43.7, 54.1, 67.7, 110.9, 117.3, 121.6, 125.5, 126.5, 128.6, 128.7, 133.4, 139.9, 140.5, 153.4, 158.4, 159.8, 160.7. Anal. Calcd. for C23H24N2O5S: C, 62.71; H, 5.49; N, 6.36. Found: C, 62.88; H, 5.61; N, 6.24.
General procedure for the synthesis of 9-bromo-pyrrolo[1,2-h][1,7]naphthyridine (7j,k) and 10-bromo-pyrido[2,3-c]pyrrolo[1,2-a]azepine (8g, h)
To a solution of derivative 7b, c and 8a, b (0.22 mmol) in anhydrous DCM (20 ml), Br2 (Sigma-Aldrich, Schnelldorf, Germany) (0.44 mmol, 0.02 mL) was added at 0 °C and the reaction mixture was stirred at room temperature for 24 h. Then the reaction mixture was evaporated under reduced pressure. The crude product was recrystallized from diethyl ether.
Ethyl 3-(benzenesulfonyl)-8-bromo-2-oxo-1,2,5,6-tetrahydropyrrolo[1,2-h][1,7]naphthyridine-8-carboxylate (7j)
This compound was obtained by reaction of 7b. Yellow solid; yield: 63%; mp: 196–197 °C; IR: 3342 (NH), 1700 (CO), 1669 (CO) cm−1; 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.37 (3H, t, J = 7.1 Hz, CH3), 2.99 (2H, t, J = 7.0 Hz, CH2), 4.33 (2H, q, J = 7.1 Hz, CH2), 4.67 (2H, t, J = 7.0 Hz, CH2), 7.03 (1H, s, Ar), 7.49–7.65 (3H, m, Ar), 8.09–8.17 (2H, m, Ar), 8.31 (1H, s, H-4), 10.02 (1H, s, NH); 13C NMR (DMSO-d6, 50 MHz, ppm): δ 14.3, 23.8, 42.8, 61.3, 97.5, 108.2, 120.8, 122.3, 126.2, 127.9, 128.7, 129.0, 133.6, 138.8, 139.6, 143.8, 156.0, 159.6. Anal. Calcd. for C20H17BrN2O5S: C, 50.32; H, 3.59; N, 5.87. Found: C, 50.16; H, 3.72; N, 5.65.
Propan-2-yl 3-(benzenesulfonyl)-8-bromo-2-oxo-1,2,5,6-tetrahydropyrrolo[1,2-h][1,7]naphthyridine-8-carboxylate (7k)
This compound was obtained by reaction of 7c. White solid; yield: 69%, mp: 174–175 °C; IR: 3336 (NH), 1705 (CO), 1669 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.40 (6H, d, J = 6.1 Hz, 2 × CH3), 3.06 (2H, s, CH2), 4.71 (2H, s, CH2), 5.20–5.28 (1H, m, CH), 7.02 (1H, s, Ar), 7.55–7.63 (3H, m, Ar), 8.17–8.19 (2H, m, Ar), 8.37 (1H, s, H-4), 9.25 (1H, s, NH); 13C NMR (CDCl3, 50 MHz, ppm): δ 22.1, 26.3, 43.7, 69.2, 98.0, 102.7, 109.3, 120.9, 122.4, 124.9, 126.8, 129.1, 129.5, 133.7, 144.6, 156.3, 158.7, 159.1. Anal. Calcd. for C21H19BrN2O5S: C, 51.33; H, 3.90; N, 5.70. Found: C, 51.09; H, 4.14; N, 5.41.
Ethyl 3-(benzenesulfonyl)-9-bromo-2-oxo-1,5,6,7-tetrahydro-2H-pyrido[2,3-c]pyrrolo[1,2-a]azepine-9-carboxylate (8g)
This compound was obtained by reaction of 8a. White solid; yield: 90%, mp: 172–173 °C; IR: 3399 (NH), 1705 (CO), 1652 (CO) cm−1; 1H NMR (CDCl3, 200 MHz, ppm): δ 1.33 (3H, t, J = 7.1 Hz, CH3), 2.18–2.21 (2H, m, CH2), 2.38–2.46 (2H, m, CH2), 4.21–4.38 (4H, m, 2 × CH2), 7.05 (1H, s, Ar), 7.58–7.75 (3H, m, Ar), 8.02 (2H, d, J = 6.9 Hz, Ar), 8.37 (1H, s, H-4), 12.48 (1H, s, NH); 13C NMR (CDCl3, 50 MHz, ppm): δ 14.0, 26.0, 31.4, 46.7, 61.1, 98.0, 106.7, 119.2, 123.0, 124.6, 128.3, 128.8, 134.0, 140.3, 140.4, 147.9, 157.3, 159.1, 159.7. Anal. Calcd. for C21H19BrN2O5S: C, 51.33; H, 3.90; N, 5.70. Found: C, 51.51; H, 3.73; N, 5.89.
Propan-2-yl 3-(benzenesulfonyl)-9-bromo-2-oxo-1,5,6,7-tetrahydro-2H-pyrido[2,3-c]pyrrolo[1,2-a]azepine-9-carboxylate (8h)
This compound was obtained by reaction of 8b. White solid; yield: 84%, mp: 167–168 °C; IR: 3422 (NH), 1702 (CO), 1646 (CO) cm−1; 1H NMR (DMSO-d6, 200 MHz, ppm): δ 1.28–1.35 (6H, m, 2 × CH3), 2.18–2.41 (2H, m, CH2), 2.44 (2H, s, CH2), 4.16 (2H, s, CH2), 5.03–5.15 (1H, m, CH), 7.00 (1H, s, H-Ar), 7.58–7.72 (3H, m, 3 × H-Ar), 8.01 (2H, d, 2H-Ar), 8.37 (1H, s, 1H-Ar), 12.11–12.81 (1H, m, NH); 13C NMR (DMSO-d6, 50 MHz, ppm): δ 22.0, 27.6, 31.8, 44.8, 68.8, 98.5, 107.5, 119.4, 119.6, 126.7, 127.7, 128.6, 129.0, 129.1, 134.0, 139.1, 142.2, 146.8, 159.2. Anal. Calcd. for C22H21BrN2O5S: C, 52.28; H, 4.19; N, 5.54. Found: C, 52.42; H, 4.01; N, 5.67.
Biology
All tested compounds were dissolved in the minimum DMSO amount prior to cell culture testing. A calculated amount of the stock DMSO solution was added to the cell culture media to reach a final maximum DMSO concentration of 0.5%, which had no discernable effects on cell viability.
Light exposure
The irradiation device used for the irradiation of cell cultures during the assays was a black box equipped with two Philips UVA lamps with an irradiance of 17 mW cm−2 and a centered emission peak at 365 nm; UVA irradiance was measured using a Waldmann Variocontrol radiometer, equipped with a UVA sensor. The box was maintained at room temperature inside with a fan and the multiwell plates were placed onto a water-refrigerated metal block.
Cell cultures
Human triple negative (TNBC) breast MDA-MB-231 carcinoma cells were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). Human embryonic kidney HEK293 cells were obtained from the European Collection of Cell Cultures (ECACC, Salisbury, UK). Cell lines were maintained in the logarithmic phase at 37 °C in a 5% CO2 atmosphere using the following culture media containing 10% foetal calf serum (Euroclone, Milan, Italy), antibiotics (50 units/mL penicillin and 50 μg/mL streptomycin) and 2 mM l-glutamine: (i) RPMI-1640 medium (Euroclone, Milan, Italy) for MDA-MB-231 4 cells; (ii) DMEM (Euroclone, Milan, Italy) for HEK293 cells.
MTT assay
Cells (5–8 × 103 cells/well, dependent upon the growth characteristics of the cell line) were seeded in 96-well microplates in growth medium (100 μL). After 24 h, the medium was removed and replaced with a fresh one containing the compound to be studied at the appropriate concentration. Triplicate cultures were established for each treatment. For photocytotoxicity studies, after 1 h of incubation the medium was replaced with PBS and cells were treated with 2.0 J cm−2 of UVA light. After irradiation, PBS was replaced with complete proliferation media. For pre-irradiation studies, instead, compounds in PBS were irradiated with 2.0 J cm−2 of UVA light and subsequently cells were treated with pre-irradiated solutions. Following 24 h, each well was treated with 10 μL of a 5 mg/mL MTT saline solution, and following 5 h of incubation, 100 μL of a sodium dodecyl sulfate (SDS) solution in HCl 0.01 M were added. After an overnight incubation, cell growth inhibition was detected by measuring the absorbance of each well at 570 nm using a Bio-Rad 680 microplate reader. Mean absorbance for each drug dose was expressed as a percentage of the control untreated well absorbance and plotted against drug concentration. IC50 values, the drug concentrations that reduce the mean absorbance at 570 nm to 50% of those in the untreated control wells, were calculated by the four-parameter logistic (4-PL) model. Evaluation was based on means from at least four independent experiments.
Cell death induction
MDA-MB-231 cells were seeded into 8-well tissue-culture slides (BD Falcon, Bedford, MA, USA) at 5 × 104 cells/well (0.8 cm2). After 24 h, the cells were washed twice with PBS and, following 24 h of treatment with IC50 doses of the tested compound, cells were stained for 5 min with 10 µg/mL of Hoechst 33258 (20-(4-hydroxyphenyl)-5-(4-methyl-1-piperazinyl)-2,50-bi-1H-benzimidazole trihydrochloride hydrate, Sigma-Aldrich, St.Louis, MI, USA) or propidium iodide (PI, Sigma-Aldrich, St.Louis, MI, USA) in PBS. Samples were examined at × 5 and × 40 magnification in a Zeiss LSM 800 confocal microscope using the Zeiss ZEN 2.3 software system.
Mitochondrial Membrane Potential (ΔΨ)
The ΔΨ was assayed using the Mito-ID® Membrane Potential Kit according to the manufacturer’s instructions (Enzo Life Sciences, Farmingdale, NY, USA) as previously described. Briefly, MDA-MB-231 cells (8 × 103 per well) were seeded in 96-well plates; after 24 h, cells were washed with PBS and loaded with Mito-ID Detection Reagent for 30 min at 37 °C in the dark. Afterwards, cells were incubated with increasing concentrations of tested compounds for 90 min and after that exposed to UVA light (2.0 J/cm2). CCCP (Carbonyl cyanide 3-chlorophenyl-hydrazone) was used as positive control. Fluorescence intensity was estimated using a VICTOR X3 (PerkinElmer, USA) plate reader at 490 (excitation) and 590 nm (emission).
ROS production
The production of ROS was measured in MDA-MB-231 cells (104 per well) grown for 24 h in a 96-well plate in RPMI medium without phenol red (Merk, Germany). Cells were then washed with PBS and loaded with 10 μM 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM–H2DCFDA) (Molecular Probes-Invitrogen, Eugene, OR) for 45 min, in the dark. Afterwards, cells were washed with PBS and incubated with tested compounds (25 µM) for 90 min and after that exposed to UVA light (2.0 J/cm2). Fluorescence increase was estimated utilizing the wavelengths of 485 nm (excitation) and 527 nm (emission) in a VICTOR X3 (PerkinElmer, USA) plate reader. Antimycin (3 μM, Merk), a potent inhibitor of Complex III in the electron transport chain, was used as positive control.
Quantification of thiols
The MDA-MB-231 cells (2 × 105) were seeded in a six-well plate in growth medium (4 mL). After 24 h, cells were incubated with tested compounds (25 µM) for 90 min, and after that exposed to UVA light (2.0 J/cm2). Subsequently, after 24 h, the thiol content was measured as previously described (Rigobello et al. 2009).
Generation of active oxygen species
Singlet oxygen determination. Samples containing the compounds under examination (2.2 × 10–5 M), p-nitrosodimethylaniline (4 × 10–5 M) and imidazole (4 × 10–5 M) in 0.02 M phosphate buffer (pH 7.3) were irradiated with increasing UVA doses and their absorbance at 440 nm was then measured (Kraljic and El Mohsni 1978). The data were expressed as percentage of RNO bleaching.
Superoxide radical determination
Samples containing the compounds under examination (10–5 M) and nitroblue tetrazolium (1.6 × 10–4 M) in 10 mM carbonate buffer (pH 10) were irradiated with increasing UVA doses, and their absorbance at 560 nm was measured (Pathak and Joshi 1984).
UVA Photostability of the compounds
Samples containing each compound (2.2 × 10− 5 M) dissolved in PBS (0.04 M) were irradiated with increasing UVA doses from 0 to 20 J/cm2 (0.2, 5, 10, 15 and 20 J/cm2), and their absorption spectrum was measured, with a Cary 50 Scan UV–Visible Spectrophotometer.
Cellular biodistribution studies
MDA-MB-231 cells were seeded into 4-well tissue-culture slides (BD Falcon, Bedford, MA, USA) at 2.5 × 104 cells/well (0.4 cm2). After 24 h, after 1 h of incubation the medium was replaced with PBS and cells were treated with 2.0 J cm−2 of UVA light. After irradiation, PBS was replaced with complete proliferation media. Samples were examined at 20 × magnification in a Zeiss LSM 800 confocal microscope using the Zeiss ZEN 2.3 software system.
Statistical analysis
Data are expressed as mean ± the standard deviation (SD) of the mean. The difference between groups was evaluated using analysis of variance (ANOVA) with Graphpad Prism 9.0.
Results
Chemistry
To access the pyrrolo[1,2-h][1,7]naphthyridinone (7) and pyrido[2,3-c]pyrrolo[1,2-a]azepinone (8) systems we started our synthesis from ketones 6,7-dihydroindolizin-8(5H)-one (n = 1) and 5,6,7,8-tetrahydro-9H-pyrrole[1,2-a]azepine-9-one (n = 2) respectively, which were properly prepared according to our methods (Barreca et al. 2022a).
The latter compounds were reacted with an excess of N,N-dimethylformamide dimethylacetal (DMFDMA) in N,N-dimethylformamide (DMF) to yield enaminoketones 9a–e in good yields (56–87%) (Scheme 1). These kinds of intermediates have been widely used by us, as they are highly reactive and prone to react with dinucleophyles furnishing several opportunities of annelation. In particular, by reaction with cyanomethylene compounds, the pyridine-2-one ring closure can be achieved. We chose to react enaminoketones 9 with phenylsulfonylacetonitrile, as the phenylsulfonyl group demonstrated crucial for the activity in our previous studies, leading to the desired tricyclic derivatives 7a–c and 8a, b (54–71%) (Scheme 1, Table 1) which were further subjected to methylation at the pyridine ring using iodomethane and NaH as the base in DMF, allowing the isolation of the corresponding N-methyl 7d–f and 8c, d (51–57%) and O-methyl substituted 7g–i and 8e, f (35–46%) derivatives from the same reaction mixtures (Scheme 1, Table 1). Additionally, compounds 7a–c and 8a, b were subjected to bromination of the pyrrole ring with the aim of evaluating the effect of the presence of bromine atom, usually responsible of the so called “heavy atom effect”, on cyto- and phototoxicity. Reactions were carried out using bromine as brominating agent, leading to pyrrolonaphthyridinones 7j, k and pyridopyrroloazepinones 8g, h (63–90%) (Scheme 1, Table 1). From the reaction of derivative 7a, it was not possible to isolate the desired bromo derivative as pure product for biological screenings.

Synthesis of pyrrolo[1,2-h][1,7]naphthyridinones (7) and pyrido[2,3-c]pyrrolo[1,2-a]azepinones (8). Reagents and conditions: (a) PhSO2CH2CN, ethanol, reflux, 24 h, 54–71%; (b) NaH, DMF, 0 °C to rt, 6 h, then iodomethane, 0 °C to rt, 24 h, 35–57%; (c) Br2, DCM, 0 °C to rt, 24 h, 63–90%
Biology
Cytotoxicity and photocitotoxicity profiles
The new synthetized pyrrolo[1,2-h][1,7]naphthyridinones 7 and pyrido[2,3-c]pyrrolo[1,2-a]azepinones 8 were screened for their cytotoxicity (in the dark) and photocitotoxicity (upon UVA light irradiation at 2.0 J/cm2) profile on human TNBC cells, MDA-MB-231. The cytotoxicity/photocytotoxicity parameters, expressed in terms of IC50 values obtained by MTT test after 24 h of drug-exposure, are listed in Table 2.
Interestingly, all compounds were completely inactive in the dark and some of them behaved as pure PSs, being effective only upon irradiation. Within the pyrrolo[1,2-h][1,7]naphthyridinone series, derivatives 7b and 7h–k showed a detectable cytotoxicity and, in particular, compound 7h and 7i elicited IC50 values in the low micromolar range (IC50 4.4 and 7.9 μM, respectively). Considering the pyrido[2,3-c]pyrrolo[1,2-a]azepinone series, however, compounds 8c and 8e–g were less effective in inhibiting cancer cell viability upon irradiation, with IC50 values in the micromolar range (IC50 15.3–24.9 μM). It is of worth noting that the O-methyl COOEt substituted derivatives 7h and 8e were the most effective compounds among the two series.
In addition, as one of the main drawbacks of chemotherapeutics is the undesired cytotoxic effect toward non-cancerous cells, we also evaluate the selectivity of the newly developed compounds against cancer cells with respect to non-cancer ones. To this attempt, compounds were screened against a human non-transformed, highly replicating cell line, the human embryonic kidney 293 (HEK293) cells. Remarkably, compounds belonging to both the pyrrolo[1,2-h][1,7]naphthyridinone and pyrido[2,3-c]pyrrolo[1,2-a]azepinone series were completely ineffective against non-cancer cells, thus attesting their preferential activity against cancer cells (Table S1).
Cell-free and in cells photogeneration of ROS
To corroborate the above reported hypothesis and to confirm the ability of all derivatives to form ROS when UVA irradiated, experiments in both cell-free and MDA-MB-231 human TNBC cells were performed. Concerning cell-free experiments, two in solution assays for singlet oxygen and superoxide anion have demonstrated to be highly predictable for many photosensitizing molecules (Kraljic and El Mohsni 1978; Pathak and Joshi 1984; Dalla Via et al. 2015; Zhang et al. 2021).
Cell-free ROS generation
With the two assays shown below, it is possible to distinguish the capacity of the test compounds of producing singlet oxygen over superoxide anion and therefore predict their behaviour as photosensitizers.
The percentage of RNO photobleaching of tested compounds under increasing light doses, which is a measure of singlet oxygen generation, is shown in Fig. 2. The majority of the compounds, with the exception of 7a and 7c, were able to generate singlet oxygen in a light dose-dependent manner (0–20 J/cm2), thus attesting that a higher UVA dose increased the singlet oxygen concentration by these compounds and, as a consequence, led to stronger oxidation of RNO/higher percentage of photobleaching. Among the two series, derivatives 7j and 7k as well as 8g and 8h were the ones with a higher percentage of RNO photobleaching, thus indicating their greater ability of singlet oxygen generation.

Variation of RNO absorbance at 440 nm as indication of singlet oxygen production by tested compounds under increasing UVA light doses (0–20 J/cm2)
Additionally, in Fig. 3 the changes in the absorbance values of NBT at 560 nm under increasing light doses are depicted. Some of the compounds only registered the production of superoxide anion under specific light dose(s) rather than under the whole light dose range. Compounds 7j, 7k, 8d, 8f, and 8h production was not reported since they did not produce superoxide anion at any light dose. Likely, superoxide anion is a free radical with a short half-life, making its measurement more difficult than singlet oxygen especially when produced in very low amount. Among all, the most effective in generating a high concentration of superoxide anion was compound 7a. Moreover, from a biological point of view, this radical species despite its high reduction potential of + 0.94 V, can oxidize very few biological compounds (D’Autréaux and Toledano 2007; Krumova et al. 2016), thus O2˙− is less effective in destroying cell components than singlet oxygen and therefore less powerful in provoking a general toxicity against treated cancer cells (see cell viability experiments).

Variation of NBT at 560 nm as indication of superoxide anion production by tested compounds under increasing UVA light doses (0–20 J/cm2)
Intracellular ROS generation
In order to understand if the intracellular action mechanism of our compounds was dependent on ROS production in intact cancer cells, the most representative compounds 7h and 8e were evaluated for their ability to increase intracellular basal ROS production in TNBC cells. With this kind of assay, all reactive oxygen species are detected without distinction among singlet oxygen, superoxide, hypochlorite, nitric oxide and hydrogen peroxide.
ROS production under light (UVA, 2.0 J/cm2) was evaluated in MDA-MB-231 cell samples treated with increasing concentrations of tested compounds or antimycin, a potent inhibitor of Complex III of mitochondria electron chain, used as a positive control under the same experimental conditions (Fig. 4).

Cellular ROS production (A) and total thiol redox state (B). A Effect of UVA activated tested compounds (25 µM) or antimycin (3 µM) on hydrogen peroxide formation in MDA-MB-231 cancer cells. B Sulfhydryl content in MDA-MB-231 cancer cells incubated for 24 h with UVA activated tested compounds. The sulfhydryl group amount was determined by the DTNB assay. Error bars indicate S.D
Treatment of MDA-MB-231 cells with 7h and 8e determined the highest increase in cellular ROS production, in a time-dependent manner (Fig. 4A). Remarkably, cells treated with 7h showed a hydrogen peroxide content comparable to that induced by antimycin. Conversely, compounds 7e and 8d were hardly effective in inducing any ROS basal level production increase.
Coherently, intracellular thiols were significantly decreased by treatment with 7h and 8e in a time-dependent manner. Interestingly, 7h was much more effective than 8e, being able to decrease intracellular sulfhydryl content of about 45% after 24 h of exposure (Fig. 4B).
Mitochondrial membrane potential loss and apoptotic cell death
Induction of ROS production can in turn prompt the collapse of mitochondrial membrane potential as well as loss of mitochondrial integrity and functioning. We hence evaluated the effect determined by treatment with representative compounds in terms of mitochondrial membrane potential changes. MDA-MB-231 TNBC cells were treated with tested compounds upon UVA irradiation and the percentage of cells with hypopolarized mitochondrial membrane potential was determined fluorometrically by means of the Mito-ID® Membrane Potential Kit.
As summarized in Fig. 5A, a significant increase of cells with depolarized mitochondria was observed after treatment with compounds 7h and 8e which was like that exerted by the positive control CCCP (Carbonyl cyanide 3-chlorophenyl-hydrazone). On the contrary, treatment with 7e and 8d did not induce a substantial change in mitochondrial membrane potential of TNBC cells.

Effects on mitochondrial membrane potential (A) and cell death induction (B) in human breast MDA-MB-231 cancer cells. A Cells were treated with 7h and 8e and stained with TMRM (10 nM). Fluorescence was estimated at 490 nm (excitation) and 590 nm (emission). Data are the means of five independent experiments. Error bars indicate S.D. * P < 0.05. B Cells were treated with concentrations of 7h and 8e equal to IC50 for 24 h and stained with the fluorescent dyes Hoechst 33342 and PI
Loss of mitochondrial transmembrane potential is a crucial step in the activation of intrinsic apoptotic cell death. Hence, the ability to induce apoptosis by representative compounds 7h and 8e was assessed in human TNBC cells. MDA-MB-231 cells were treated with IC50 doses of tested compounds and apoptotic cell death induction was confirmed through a Hoechst 33342 staining assay.
As reported in Fig. 5B, cells treated with 7h and 8e, as compared with control untreated cells, were positive to Hoechst 33342 staining, and displayed chromatin condensation and fragmentation as well as pyknotic nuclei, typical features of apoptosis. On the other hand, treated cells were poor stained with PI, thus indicating the absence of necrosis induction.
Photostability experiments
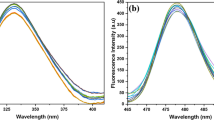
The most significant 7h and 8e pyridone derivatives were tested for their stability under UVA irradiation. These data might be useful to understand the mechanism of action of the compounds apart from the production of ROS. As shown by the changes of their UV–Vis spectra under increasing UVA doses (Fig. 6), compound taken in the dark = 0 J/cm2 = red spectrum; compound under 20 J/cm2 of UVA light = light green spectrum), the tested compounds were very unstable under light and behaved very similarly. Indeed, their main absorption peak gradually decreased under irradiation accompanied by a blue shift of the maximum and the change of its shape, suggesting the loss of the original structure. Moreover, isosbestic points were visible, indicating the formation of one or more new stable species in equilibrium with the intact dark compound.

UV–Vis Absorption spectra of 7h (left) and 8e (right) in PBS irradiated under 0, 2, 5, 10, 15 and 20 J/cm2 with UVA light. 0 J/cm2, red spectrum; 20 J/cm2, light green spectrum
In addition, to test the role of the photodegradation products on the photocytotoxicity profiles of the most representative derivatives 7h and 8e, the cytotoxic activity of the photodegraded mixture was evaluated by treating TNBC cells with the corresponding pre-irradiated (20 J/cm2) PBS solutions. No cytotoxic activity was evidenced (IC50 over 50 µM) clearly attesting that the photodegradation products did not frankly contribute to the cytotoxic activity of the test compounds; rather, these compounds lose their activity when photodegraded and suggest that they mainly act by inducing intracellular ROS.
By roughly calculating the decrease of absorbance of the main UV–Vis peaks (data non shown), the photodegradation was very weak under the dose of light used during the cytotoxic experiments (2 J/cm2, blue line in Fig. 5) and the yield of photoproducts formed able to participate to the activity of 7h and 8e was most likely not significant for their contribution in their general cytotoxicity. Anyway, even in very high amount (after 20 J/cm2 of UVA, light green line in Fig. 5), they were not active at all as discussed above.
Cellular accumulation and biodistribution studies
With the aim of correlating cellular phototoxic activity with intracellular accumulation of tested compounds, we performed some preliminary in cell biodistribution studies. By exploiting their fluorescence characteristics, the most effective compounds 7h and 8e, as well as a completely ineffective derivative, namely 8h, were evaluated for their ability to enter MDA-MB-231 TNBC cells and localize into different cellular compartments by confocal microscopy analysis.
As evident form micrographs reported in Fig. 7, the most photocytotoxic compounds 7h and 8e were able to significantly enter cancer cell membrane and accumulate in cytoplasm. Compounds preferentially localize in the perinuclear space, with minimal accumulation in the periplasmatic site. Conversely, compound 8h was barely effective in accumulating into cancer cells, and only low levels of fluorescence were detected in the cytoplasm of treated cancer cells.

Intracellular accumulation studies. MDA-MB-231 TNBC cells were treated with 7h, 8e and 8h upon irradiation and live samples were examined at × 20 magnification by a confocal microscope
Overall, these cellular accumulation studies clearly support the hypothesis that the cell killing inactivity of compound 8h can be explained by taking into consideration its scarce efficacy to permeate TNBC cells. Despite behaving as PS in cell-free experiments, 8h was found completely ineffective in inhibiting cancer cell proliferation upon irradiation.
Discussion
Photochemotherapy broadly refers to a treatment method that uses a photosensitizer (PS) activated by a selected light irradiation. The combination of both produces a better effect than the use of one of these treatments alone (Ledo and Ledo 2000). This therapeutic technique comprises PUVA (psoralen-UV-A) therapy, in which a psoralen derivative damages DNA of proliferating cells under the exposure to ultraviolet A (UV-A) radiation (Schneider et al. 2008) and photodynamic therapy (PDT) that relies on the red-light activation of a porphyrin-like derivative that generates reactive oxygen species leading to the destruction of unwanted cells and diseased tissues (Benov 2015). The lack of toxicity of the above photosensitizers in the dark and the selective illumination of the targeted area avoids the general toxicity that is one of the main drawbacks of the conventional chemotherapy against cancer. Therefore, the ideal PS would be a stable drug, with specific uptake by the targeted tissues, minimal dark cytotoxicity, strong absorption at relatively long wavelengths (600–800 nm) at which the light can penetrate efficiently into the tissues, rapid clearance to avoid phototoxic side effects, being amphiphilic, chemically pure and easy to access or synthesize (Dias et al. 2021). In the case of PS activated by less penetrating shorter wavelengths, such as psoralen derivatives activated by UVA light and, as demonstrated more recently, even by blue light (Sturaro et al. 2021), their application against proliferating cells contemplates the treatment of skin diseases, i.e. psoriasis and neurodermitis, or an extracorporeal treatment, the so called extracorporeal photopheresis (ECP) against some immunological disorders, i.e., cutaneous T cell lymphoma and graft versus host disease (Drexler et al. 2020). Although a big progress has been done towards the understanding of TNBC’s biology and the development of more efficient treatment regimens, drug resistance, strong side effects and episodes of recurrence are still common among patients with this aggressive disease. Intensive research has been conducted to uncover a new therapeutical approach which maintains a patient’s quality of life and has a more localized action. Quinazoline derivatives arose from this diligent search as a promising direction in the treatment of this life- threatening cancer disease. Their enormous potential is the motivation for this study, whose aims consisted of testing the photocytotoxic activity of nineteen newly synthesised photosensitizing pyridone derivatives on a TNBC cancer cell line (MDA-MB-231) under UV-A light and study their ability to produce ROS as the main mechanism of their action.
The demonstrated antiproliferative activity in the micromolar range of some of these compounds in combination with a small non-toxic dose of UVA light (2.0 J/cm2), their inactivity in the dark against both normal and cancer cells, their apoptotic mechanism of cell death make some of these compounds as a very promising alternative to the conventional and often not effective therapies against TNBC. Interestingly, all the compounds presenting a bromine as substituent at the pentatomic ring were able to generate high yields of singlet oxygen. This is in line with the effect of the heavy atom in the molecular structure which increases the spin orbit coupling between singlet and triplet states, leading to more efficient ISC with respect to the non-halogenated compounds (Lai et al. 2021). Unfortunately, these compounds bearing the heavy halogen bromine atom as inducer of singlet oxygen production could not enter the cells so efficiently and their potential higher activity with respect the other compounds was lost in the presence of cancer cells. Indeed, the localization of the compounds inside the cells confirmed this assumption.
It must be noted that, as in the case of PUVA, we evaluated the toxicity of these compounds under UVA light, where all absorbed significantly; but unfortunately, UVA light penetrates the tissues much less than the red light used in PDT. Theoretically, PUVA therapy could not be used to treat solid internal cancers or deep skin cancers as PDT. However, a direct irradiation of the diseased area in the presence of our compounds to get a clean tissue during and after the surgical removal of the tumour could be suggested. Moreover, for superficial breast cancers the use of a catheter for the instillation of the photoactive principle followed by irradiation through a probe, equipped with a tip emitting UVA light, could be another option for these potential phototherapeutic compounds.
References
Ali S, Buczek D, Jassem J (2020) Changing paradigms in breast cancer treatment. Eur J Transl Clin Med 3:53–63. https://doi.org/10.31373/ejtcm/130486
Barraja P, Diana P, Montalbano A, Dattolo G, Cirrincione G, Viola G, Vedaldi D, Dall’Acqua F (2006) Pyrrolo[2,3-h]quinolinones: a new ring system with potent photoantiproliferative activity. Bioorg Med Chem 14:8712–8728. https://doi.org/10.1016/j.bmc.2006.07.061
Barraja P, Caracausi L, Diana P, Carbone A, Montalbano A, Cirrincione G, Brun P, Castagliuolo I, Dall’Acqua F, Vedaldi D, Salvador A (2010) Synthesis of pyrrolo[3,2-h]quinolinones with good photochemotherapeutic activity and no DNA damage. Bioorg Med Chem 18:4830–4843. https://doi.org/10.1016/j.bmc.2010.04.080
Barraja P, Diana P, Montalbano A, Carbone A, Viola G, Basso G, Salvador A, Vedaldi D, Dall’Acqua F, Cirrincione G (2011) Pyrrolo[3,4-h]quinolinones a new class of photochemotherapeutic agents. Bioorg Med Chem 19:2326–2341. https://doi.org/10.1016/j.bmc.2011.02.023
Barreca M, Ingarra AM, Raimondi MV, Spanò V, De Franco M, Menilli L, Gandin V, Miolo G, Barraja P, Montalbano A (2022a) Insight on pyrimido[5,4-g]indolizine and pyrimido[4,5-c]pyrrolo[1,2-a]azepine systems as promising photosensitizers on malignant cells. Eur J Med Chem 237:114399. https://doi.org/10.1016/j.ejmech.2022.114399
Barreca M, Spanò V, Raimondi MV, Bivacqua R, Giuffrida S, Montalbano A, Cavalli A, Bertoni F, Barraja P (2022b) GPCR inhibition in treating lymphoma. ACS Med Chem Lett 13:358–364. https://doi.org/10.1021/acsmedchemlett.1c00600
Benov L (2015) Photodynamic therapy: current status and future directions. Med Princ Pract 24:14–28. https://doi.org/10.1159/000362416
Bergin ART, Loi S (2019) Triple-negative breast cancer: recent treatment advances [version 1; peer review: 2 approved]. F1000Research 8:1342. https://doi.org/10.12688/f1000research.18888.1
Cilibrasi V, Spanò V, Bortolozzi R, Barreca M, Raimondi MV, Maruca A, Montalbano A, Alcaro S, Ronca R, Viola G, Barraja P (2022) Synthesis of 2H-Imidazo[2′,1’:2,3] [1,3]thiazolo[4,5-e]isoindol-8-yl-phenylureas with promising therapeutic features for the treatment of acute myeloid leukemia (AML) with FLT3/ITD mutations. Eur J Med Chem 235:114292. https://doi.org/10.1016/j.ejmech.2022.114292
Criscitiello C, Azim HA, Schouten PC, Linn SC, Sotiriou C (2012) Understanding the biology of triple-negative breast cancer. Ann Oncol 23:13–18. https://doi.org/10.1093/annonc/mds188
D’Autréaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8:813–824. https://doi.org/10.1038/nrm2256
Dalla Via L, Marzaro G, Mazzoli A, Chilin A, Miolo G (2015) Photobiological properties of 3-psoralenacetic acids. Photochem Photobiol Sci 14:2074–2086. https://doi.org/10.1039/c5pp00210a
Dias CJ, Helguero L, Faustino MAF (2021) Current photoactive molecules for targeted therapy of triple-negative breast cancer. Molecules 26:7654. https://doi.org/10.3390/molecules26247654
Drexler B, Buser A, Infanti L, Stehle G, Halter J, Holbro A (2020) Extracorporeal photopheresis in Graft-versus-host disease. Transfus Med Hemother 47:214–224. https://doi.org/10.1159/000508169
Kraljic I, El Mohsni S (1978) A new method for the detection of singlet oxygen in acqueosus solutions. Photochem Photobiol 28:577–581. https://doi.org/10.1111/j.1751-1097.1978.tb06972.x
Krumova K, Cosa G, Aubry J, Kanofsky JR (2016) Section I: Fundamentals. In: Singlet oxygen : applications in biosciences and nanosciences, pp 1–2. https://doi.org/10.1039/9781782622208-00001
Labbozzetta M, Barreca M, Spanò V, Raimondi MV, Poma P, Notarbartolo M, Barraja P, Montalbano A (2022) Novel insights on [1,2]oxazolo[5,4-e]isoindoles on multidrug resistant acute myeloid leukemia cell line. Drug Dev Res. https://doi.org/10.1002/ddr.21962
Lai L, Fang B, Fan M, Cheng W, Meizhen Y (2021) Modulating room-temperature phosphorescence through the synergistic effect of heavy-atom effect and halogen bonding. J Phys Chem C 125:16350–16357. https://doi.org/10.1021/acs.jpcc.1c04989
Ledo E, Ledo A (2000) Phototherapy, photochemotherapy, and photodynamic therapy: unapproved uses or indications. Clin Dermatol 18:77–86. https://doi.org/10.1016/s0738-081x(99)00096-6
Liu J, Ming B, Gong GH, Wang D, Bao GL, Yu LJ (2018) Current research on anti-breast cancer synthetic compounds. RSC Adv 8:4386–4416. https://doi.org/10.1039/c7ra12912b
Lüönd F, Tiede S, Christofori G (2021) Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during malignant progression. Br J Cancer 125:164–175. https://doi.org/10.1038/s41416-021-01328-7
Miolo G, Salvador A, Mazzoli A, Spallett A, Marzaro G, Chilin A (2014) Photochemical and photobiological studies on furoquinazolines as new psoralen analogs. J Photochem Photobiol B 138:43–54. https://doi.org/10.1016/j.jphotobiol.2014.05.002
Pathak MA, Joshi PC (1984) Production of active oxygen species (1O2 and O2⨪) by psoralens and ultraviolet radiation (320–400 nm). BBA 798:115–126. https://doi.org/10.1016/0304-4165(84)90018-7
Rigobello MP, Gandin V, Folda A, Rundlof AK, Fernandes AP, Bindoli A, Marzano C, Bjornstedt M (2009) Treatment of human cancer cells with selenite or tellurite in combination with auranofin enhances cell death due to redox shift. Free Radic Biol Med 47:710–721. https://doi.org/10.1016/j.freeradbiomed.2009.05.027
Schneider LA, Hinrichs R, Scharffetter-Kochanek K (2008) Phototherapy and photochemotherapy. Clin Dermatol 26:464–476. https://doi.org/10.1016/j.clindermatol.2007.11.004
Spanò V, Giallombardo D, Cilibrasi V, Parrino B, Carbone A, Montalbano A, Frasson I, Salvador A, Richter SN, Doria F, Freccero M, Cascioferro S, Diana P, Cirrincione G, Barraja P (2017) Pyrrolo[3′,2′:6,7]cyclohepta[1,2-b]pyridines with potent photo-antiproliferative activity. Eur J Med Chem 128:300–318. https://doi.org/10.1016/j.ejmech.2017.02.008
Spanò V, Barreca M, Cilibrasi V, Genovese M, Renda M, Montalbano A, Galietta LJV, Barraja P (2021a) Evaluation of fused pyrrolothiazole systems as correctors of mutant CFTR protein. Molecules 26:1–25. https://doi.org/10.3390/molecules26051275
Spanò V, Barreca M, Rocca R, Bortolozzi R, Bai R, Carbone A, Raimondi MV, Palumbo Piccionello A, Montalbano A, Alcaro S, Hamel E, Viola G, Barraja P (2021b) Insight on [1,3]thiazolo[4,5-e]isoindoles as tubulin polymerization inhibitors. Eur J Med Chem 212:113122. https://doi.org/10.1016/j.ejmech.2020.113122
Sporikova Z, Koudelakova V, Trojanec R, Hajduch M (2018) Genetic markers in triple-negative breast cancer. Clin Breast Cancer 18:e841–e850. https://doi.org/10.1016/j.clbc.2018.07.023
Sturaro G, Tasso A, Menilli L, Di Liddo R, Miolo G, Conconi MT (2021) 4,6,4′-trimethylangelicin photoactivated by blue light might represent an interesting option for photochemotherapy of non-invasive bladder carcinoma: an in vitro study on T24 cells. Biomolecules 11:1–14. https://doi.org/10.3390/biom11020158
Tong CWS, Wu M, Cho WCS, To KKW (2018) Recent advances in the treatment of breast cancer. Front Oncol 8:227. https://doi.org/10.3389/fonc.2018.00227
Yao H, He G, Yan S, Chen C, Song L, Rosol TJ, Deng X (2017) Triple-negative breast cancer: is there a treatment on the horizon? Oncotarget 8:1913–1924. https://doi.org/10.18632/oncotarget.12284
Zhang Z, Fan J, Du J, Peng X (2021) Two-channel responsive luminescent chemosensors for dioxygen species: Molecular oxygen, singlet oxygen and superoxide anion. Coord Chem Rev 427:213575. https://doi.org/10.1016/j.ccr.2020.213575
Acknowledgements
This work was financially supported by Ministero dell’Università e della Ricerca (MUR).
Funding
Open access funding provided by Università degli Studi di Palermo within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barreca, M., Ingarra, A.M., Raimondi, M.V. et al. New tricyclic systems as photosensitizers towards triple negative breast cancer cells. Arch. Pharm. Res. 45, 806–821 (2022). https://doi.org/10.1007/s12272-022-01414-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-022-01414-1