
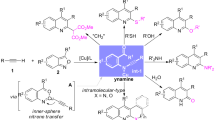
Abstract
The site-selective and metal-free C–H nitration reaction of quinoxalinones and pyrazinones as biologically important N-heterocycles with t-butyl nitrite is described. A wide range of quinoxalinones were efficiently applied in this transformation, providing C7-nitrated quinoxalinones without undergoing C3-nitration. From the view of mechanistic point, the radical addition reaction exclusively occurred at the electron-rich aromatic region beyond electron-deficient N-heterocycle ring. This is a first report on the C7–H functionalization of quinoxalinones under metal-free conditions. In contrast, the nitration reaction readily takes place at the C3-position of pyrazinones. This transformation is characterized by the scale-up compatibility, mild reaction conditions, and excellent functional group tolerance. The applicability of the developed method is showcased by the selective reduction of NO2 functionality on the C7-nitrated quinoxalinone product, providing aniline derivatives. Combined mechanistic investigations aided the elucidation of a plausible reaction mechanism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the landmark discovery by Mitscherlich and Laurent in 1834 (Patel et al. 2021), nitro compounds are important and versatile building blocks in organic chemistry (Ono 2001), and their derivatives are widely utilized in various pharmaceuticals, agrochemicals, pigments, and dyes as well as a variety of fine chemicals such as solvents, perfumes, explosives, and polymers, as shown in Fig. 1 (Fan et al. 2004; McNamara et al. 2011; Nepali et al. 2019). For example, chloramphenicol (Rebstock et al. 1949) and metronidazole (Freeman et al. 1997) are well-known antibiotic and antiprotozoal drugs for the treatment of a number of infectious diseases. The biological properties of these molecules are closely related with (hetero)aryl motifs tethered with nitro functionality, but vary depending on the nature and position of substituents on (hetero)aryl rings.

Nitro-containing pharmaceuticals and functional materials
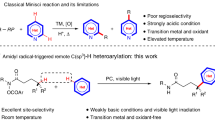
Traditional methods for nitration reactions rely on an excess use of nitric acid or its mixture with sulfuric acid or dinitrogen pentoxide (Olah et al. 1990). However, from a synthetic point of view, these protocols represent the limited functional group tolerance and the generation of undesirable by-products as well as incomplete regioselectivity. To overcome these limitations, new nitrating agents such as nitrate (Manna et al. 2012; Zolfigol et al. 2012), nitrite salts (Fors et al. 2009; Li et al. 2013), and tert-butyl nitrite (TBN) have been intensively investigated (Wu and Neumann et al. 2011; Wu and Schranck et al. 2011; Shen et al. 2014).
With great advance on C–H functionalization reactions (Mishra et al. 2017, 2018; Pandey et al. 2018; Sambiagio et al. 2018; Lee et al. 2019), direct C–H nitration of (hetero)arenes has been recently developed. The metal-free oxidative C–H nitration of phenols or amines has been explored, as shown in Fig. 2 (Koley et al. 2009; Kilpatrick et al. 2013; Li et al. 2014). However, none of these protocols represent a general strategy to allow for complete site-selectivity between the ortho- and para-positions. A great deal of effort on site-selectivity of nitration has been devoted to the transition-metal-catalyzed ortho-C–H nitration of N-heterocycles. For example, Liu reported the Pd(II)-catalyzed ortho-C–H nitration of nitrogen-containing heterocycles with silver nitrite in the presence of K2S2O8 as an external oxidant (Liu et al. 2010). The ortho-C–H nitration of (hetero)arenes using nitrite salts was also realized with the Cu(II), Rh(III), Ru(0), and Ni(II) catalytic systems (Zhang et al. 2011; Xie et al. 2013; Katayev et al. 2014; Majhi et al. 2014; Fan and Ni 2016; Wan et al. 2017). In addition, the Pd(II)-catalyzed aerobic oxidative ortho-C–H nitration of arenes with tert-butyl nitrite and toluene as the radical precursors was demonstrated (Liang et al. 2015). The azaindole-assisted ortho-C–H nitration of arenes with tert-butyl nitrite affording various nitrated azaindole derivatives was disclosed (Chun et al. 2018).

C–H nitration methods of (hetero)arenes using nitrating agents
Despite the compelling progress on the C–H nitration reaction of various N-heterocycles, the site-selective nitration reaction of quinoxalinones and pyrazinones under milder reaction conditions is still unexplored. Driven by our ongoing interest in the C−H functionalization of N-heterocycles (Han et al. 2018; Ghosh et al. 2019, 2021; An et al. 2020; Park et al. 2021), we herein describe the metal-free and site-selective C–H nitration reaction of quinoxalinones and pyrazinones with tert-butyl nitrite as a readily available nitrating agent. Notably, the gram-scale reaction, selective reduction of a nitro group, and thiocarbonylation demonstrate the synthetic utility of the developed method.
Materials and methods
General methods
Commercially available reagents were used without additional purification, unless otherwise stated. Quinoxalinones (1a–1m) and 5-aryl pyrazinones (4a–4j) were prepared according to the reported literature (Ghosh et al. 2021; Guo et al. 2021). t-Butyl nitrite was purchased from Aldrich, Switzerland. All the reactions were performed in an oil bath by using hot plate magnetic stirrer (IKA universal, Guangzhou city, China). Sealed tubes were purchased from Fischer Scientific (13 × 100 mm, 1495925A; Mexico) and dried in oven for overnight and cooled at room temperature prior to use. Thin layer chromatography was carried out using plates coated with silica gel 60 F254 (Merck KGaA, 64271 Darmstadt, Germany). For flash column chromatography, silica gel 60 Å (230–400 mesh, Merck, Germany) was used. Nuclear magnetic resonance spectra (1H, 13C, and 19F NMR) were recorded on a Bruker Unity 400, 500, and 700 MHz spectrometers in CDCl3, CD3COCD3, and DMSO-d6 solution and chemical shifts are reported as parts per million (ppm). Resonance patterns are reported with the notations s (singlet), br (broad), d (doublet), t (triplet), q (quartet), sext (sextet), dd (doublet of doublets), dt (doublet of triplets), dq (doublet of quartets), qd (quartet of doublets), td (triplet of doublets), tt (triplet of triplets), and m (multiplet). In addition, the notation br is used to indicate a broad signal. Coupling constants (J) are reported in hertz (Hz). IR spectra were recorded on a Varian 2000 Infrared spectrophotometer and are reported as cm−1. High-resolution mass spectra (HRMS) were recorded on a JEOL JMS-600 spectrometer.
General procedure and characterization data for the C7-nitration of quinoxalinones (3a–3m)
To an oven-dried sealed tube charged with 1-methylquinoxalin-2(1H)-one (1a) (32.0 mg, 0.2 mmol, 100 mol %) was added t-butyl nitrite (2a) (71.4 µL, 0.6 mmol, 300 mol %) and CH3CN (2 mL) under O2 atmosphere at room temperature. After using O2 balloon, the reaction mixture was allowed to stir at 60 °C for 20 h. The reaction mixture was cooled to room temperature, diluted with EtOAc (4 mL) and concentrated in vacuo. The residue was purified by flash column chromatography (n-hexanes/EtOAc = 10:1 to 2:1) to afford 3a (31.2 mg) in 76% yield.
1-Methyl-7-nitroquinoxalin-2(1H)-one (3a)
31.2 mg (76%); eluent (n-hexanes/EtOAc = 10:1 to 2:1); brown solid; mp = 228.9–231.1 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H), 8.32 (d, J = 2.4 Hz, 1H), 8.16 (dd, J = 8.8, 2.4 Hz, 1H), 8.06 (d, J = 8.8 Hz, 1H), 3.68 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.1, 153.9, 147.9, 136.0, 133.9, 130.9, 117.7, 110.6, 28.9; IR (KBr) υ 2924, 2854, 1666, 1587, 1512, 1462, 1354 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C9H7N3O3 205.0487; Found 205.0486.
1-Ethyl-7-nitroquinoxalin-2(1H)-one (3b)
32.9 mg (75%); eluent (n-hexanes/acetone = 10:1 to 1:1); brown solid; mp = 165.5–167.9 °C; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 8.24 (d, J = 2.4 Hz, 1H), 8.18 (dd, J = 8.8, 2.4 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 4.36 (q, J = 7.2 Hz, 2H), 1.43 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 154.1, 153.9, 148.7, 136.9, 132.8, 132.1, 118.1, 109.7, 37.8, 12.6; IR (KBr) υ 2989, 2924, 1668, 1589, 1523, 1471, 1442, 1344, 1244, 1103 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C10H9N3O3 219.0644; Found 219.0639.
1-Isobutyl-7-nitroquinoxalin-2(1H)-one (3c)
36.6 mg (74%); eluent (n-hexanes/acetone = 10:1 to 1:1); light brown solid; mp = 121.8–124.0 °C; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 8.21 (d, J = 2.4 Hz, 1H), 8.15 (dd, J = 8.8, 2.4 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 4.15 (d, J = 7.6 Hz, 2H), 2.27 (sep, J = 6.4 Hz, 1H), 1.04 (s, 3H), 1.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.8, 153.9, 148.5, 136.9, 133.4, 132.0, 118.1, 110.3, 49.3, 27.4, 20.2; IR (KBr) υ 3114, 2956, 2927, 2871, 1664, 1591, 1562, 1522, 1464, 1441, 1338, 1315, 1236, 1132, 1099, 1057 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C12H13N3O3 247.0957; Found 247.0954.
1-Butyl-7-nitroquinoxalin-2(1H)-one (3d)
35.2 mg (71%); eluent (n-hexanes/acetone = 10:1 to 3:1); yellow solid; mp = 112.3–114.3 °C; 1H NMR (400 MHz, CDCl3) δ 8.40 (s, 1H), 8.22 (d, J = 2.0 Hz, 1H), 8.16 (dd, J = 8.8, 2.0 Hz, 1H), 8.03 (d, J = 8.4 Hz, 1H), 4.28 (dd, J = 6.0 Hz, 2H), 1.80–1.73 (m, 2H), 1.51 (sext, J = 7.2 Hz, 2H), 1.02 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 154.3, 153.8, 148.6, 136.9, 133.1, 132.0, 118.1, 109.9, 42.5, 29.5, 20.3, 13.8; IR (KBr) υ 2956, 2925, 1668, 1593, 1527, 1462, 1346, 1265 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C12H13N3O3 247.0957; Found 247.0954.
1-(4-Bromobutyl)-7-nitroquinoxalin-2(1H)-one (3e)
51.2 mg (78%); eluent (n-hexanes/acetone = 10:1 to 1:1); yellow solid; mp = 126.3–128.5 °C; 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 8.26 (d, J = 2.4 Hz, 1H), 8.18 (dd, J = 8.4, 2.0 Hz, 1H), 8.05 (d, J = 8.4 Hz, 1H), 4.33 (t, J = 7.6 Hz, 2H), 3.50 (t, J = 5.6 Hz, 2H), 2.08–1.95 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 154.3, 153.7, 148.7, 136.9, 132.9, 132.2, 118.3, 109.7, 41.6, 32.6, 29.6, 25.9; IR (KBr) υ 2922, 2854, 1666, 1593, 1564, 1525, 1444, 1344, 1317, 1103 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C12H12BrN3O3 325.0062; Found 325.0061.
1-Benzyl-7-nitroquinoxalin-2(1H)-one (3f)
30.9 mg (55%); eluent (n-hexanes/EtOAc = 10:1 to 2:1); light brown solid; mp = 165.7–167.9 °C; 1H NMR (400 MHz, CDCl3) δ 8.52 (s, 1H), 8.23 (d, J = 2.0 Hz, 1H), 8.12 (dd, J = 8.8, 2.0 Hz, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.38–1.28 (m, 5H), 5.51 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 154.6, 153.9, 148.5, 136.9, 134.2, 133.1, 131.9, 129.5, 128.5, 127.3, 118.4, 110.6, 46.1; IR (KBr) υ 2922, 2854, 1666, 1593, 1564, 1523, 1450, 1342, 1317, 1219, 1103 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C15H11N3O3 281.0800; Found 281.0800.
3-((7-Nitro-2-oxoquinoxalin-1(2H)-yl)methyl)benzonitrile (3g)
19.2 mg (31%); eluent (n-hexanes/acetone = 10:1 to 2:1); yellow solid; mp = 193.8–196.1 °C; 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 8.17 (dd, J = 5.2, 1.2 Hz, 1H), 8.09 (d, J = 0.8 Hz, 1H), 8.08 (d, J = 2.8 Hz, 1H), 7.61 (dt, J = 4.4 Hz, 1H), 7.60–7.58 (m, 1H), 7.54–7.53 (m, 1H), 7.51 (t, J = 4.4 Hz, 1H), 5.53 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 154.4, 153.7, 148.7, 136.9, 135.8, 132.8, 132.4, 132.3, 131.7, 130.6, 130.3, 118.8, 118.1, 113.9, 109.9, 45.4; IR (KBr) υ 2924, 2854, 2231, 1670, 1593, 1566, 1525, 1448, 1344 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C16H10N4O3 306.0753; Found 306.0748.
1-(4-Ethoxyphenyl)-7-nitroquinoxalin-2(1H)-one (3h)
34.4 mg (56%); eluent (n-hexanes/EtOAc = 10:1 to 2:1); orange solid; mp = 182.0–183.9 °C; 1H NMR (700 MHz, CD3COCD3) δ 8.40 (s, 1H), 8.14 (dd, J = 8.4, 2.1 Hz, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.56 (d, J = 2.8 Hz, 1H), 7.39 (dt, J = 9.1, 2.8 Hz, 2H), 7.20 (dt, J = 9.1, 3.5 Hz, 2H), 4.18 (q, J = 7.0 Hz, 2H), 1.44 (t, J = 7.0 Hz, 3H); 13C NMR (175 MHz, CD3COCD3) δ 160.9, 155.6, 155.1, 149.1, 137.3, 136.6, 131.9, 130.5, 128.2, 118.4, 116.8, 111.8, 64.6, 15.1; IR (KBr) υ 2925, 2854, 1676, 1593, 1523, 1508, 1477, 1435, 1344, 1302, 1246, 1043 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C16H13N3O4 311.0906; Found 311.0904.
7-Nitro-1-(4-nitrophenyl)quinoxalin-2(1H)-one (3i)
18.8 mg (30%); eluent (n-hexanes/EtOAc = 10:1 to 3:1); yellow solid; mp = 244.2–245.6 °C; 1H NMR (400 MHz, CDCl3) δ 8.55 (dt, J = 8.8, 2.8 Hz, 2H), 8.52 (s, 1H), 8.20 (dd, J = 8.8, 2.4 Hz, 1H), 8.12 (d, J = 8.8 Hz, 1H), 7.58–7.54 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 154.0, 153.6, 148.9, 148.6, 139.6, 136.3, 133.6, 132.1, 129.8, 126.3, 119.2, 110.7; IR (KBr) υ 3111, 3082, 2924, 1680, 1595, 1523, 1435, 1348 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C14H8N4O5 312.0495; Found 312.0490.
7-Nitro-1-(m-tolyl)quinoxalin-2(1H)-one (3j)
33.8 mg (60%); eluent (n-hexanes/EtOAc = 10:1 to 3:1); yellow solid; mp = 183.1–185.6 °C; 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 8.14 (dd, J = 8.8, 2.4 Hz, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.60 (d, J = 2.4 Hz, 1H), 7.56 (td, J = 8.0, 1.6 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.10–7.08 (m, 2H), 2.47 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.4, 154.3, 148.5, 141.4, 136.3, 134.7, 134.1, 131.5, 130.8, 128.5, 124.9, 119.9, 118.5, 111.6, 21.6; IR (KBr) υ 2924, 2854, 1678, 1593, 1562, 1527, 1433, 1344 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C15H11N3O3 281.0800; Found 281.0803.
Ethyl 3-(7-nitro-2-oxoquinoxalin-1(2H)-yl)benzoate (3k)
29.2 mg (43%); eluent (n-hexanes/acetone = 10:1 to 2:1); yellow oil; 1H NMR (400 MHz, CD3COCD3) δ 8.45 (s, 1H), 8.28 (dt, J = 7.6, 1.6 Hz, 1H), 8.19–8.12 (m, 3H), 7.88 (t, J = 7.6 Hz, 1H), 7.81 (dq, J = 7.6, 1.2 Hz, 1H), 7.47 (d, J = 2.4 Hz, 1H), 7.47 (qd, J = 7.2, 1.6 Hz, 2H), 1.36 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 165.7, 155.5, 154.9, 149.1, 137.2, 136.4, 135.9, 134.1, 133.9, 132.1, 131.7, 131.6, 130.6, 118.7, 111.5, 62.0, 14.5; IR (KBr) υ 3066, 2924, 2854, 1716, 1680, 1593, 1562, 1525, 1436, 1344, 1269, 1211, 1182, 1103, 1082, 1022 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C17H13N3O5 339.0855; Found 339.0852.
6-Bromo-1-methyl-7-nitroquinoxalin-2(1H)-one (3l)
19.9 mg (35%); eluent (n-hexanes/EtOAc = 10:1 to 2:1); light brown solid; mp = 229.4–231.2 °C; 1H NMR (400 MHz, CD3COCD3) δ 8.33 (s, 1H), 8.24 (s, 1H), 8.16 (s, 1H), 3.72 (s, 3H); 13C NMR (100 MHz, CD3COCD3) δ 155.3, 154.9, 147.8, 135.9, 134.9, 112.8, 106.2, 23.3; IR (KBr) υ 2922, 2852, 1664, 1585, 1554, 1533, 1456, 1344 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C9H6BrN3O3 282.9593; Found 282.9593.
7-Chloro-1-methyl-5-nitroquinoxalin-2(1H)-one (3m)
21.6 mg (45%); eluent (n-hexanes/EtOAc = 10:1 to 3:1); light brown solid; mp = 229.4–231.5 °C; 1H NMR (700 MHz, CD3COCD3) δ 8.24 (s, 1H), 7.91 (d, J = 1.2 Hz, 1H), 7.83 (d, J = 1.2 Hz, 1H), 3.74 (s, 3H); 13C NMR (175 MHz, CD3COCD3) δ 154.9, 153.5, 150.2, 137.0, 136.7, 124.3, 118.3, 117.4, 23.3; IR (KBr) υ 2918, 2861, 1676, 1603, 1537, 1454, 1379, 1267 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C9H6ClN3O3 239.0098; Found 239.0096.
General procedure and characterization data for the C3-nitration of 5-aryl pyrazinones (5a–5j)
To an oven-dried sealed tube charged with 1-methyl-5-phenylpyrazin-2(1H)-one (4a) (37.2 mg, 0.2 mmol, 100 mol %) was added t-butyl nitrite (2a) (119.0 µL, 1.0 mmol, 500 mol %) and CH3CN (2.5 mL) under O2 atmosphere at room temperature. After using O2 balloon, the reaction mixture was allowed to stir at 60 °C for 28 h. The reaction mixture was cooled to room temperature, diluted with EtOAc (4 mL) and concentrated in vacuo. The residue was purified by flash column chromatography (n-hexane/acetone = 10:1 to 3:1) to afford 5a (33.4 mg) in 72% yield.
1-Methyl-3-nitro-5-phenylpyrazin-2(1H)-one (5a)
33.4 mg (72%); eluent (n-hexane/acetone = 10:1 to 3:1); yellow solid; mp = 191.2–193.8 °C; 1H NMR (700 MHz, DMSO-d6) δ 8.93 (s, 1H), 7.83–7.81 (m, 2H), 7.50–7.47 (m, 2H), 7.39 (tt, J = 7.7, 0.7 Hz, 1H), 3.69 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 148.4, 148.1, 135.9, 133.6, 128.9, 128.4, 127.9, 124.6, 38.3; IR (KBr) υ 3060, 2927, 1674, 1604, 1543, 1493, 1419, 1267 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C11H9N3O3 231.0644; Found 231.0641.
1-Methyl-3-nitro-5-(4-(trifluoromethoxy)phenyl)pyrazin-2(1H)-one (5b)
32.2 mg (51%); eluent (n-hexanes/acetone = 10:1 to 3:1); yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.90 (s, 1H), 7.74 (dt, J = 7.2, 2.4 Hz, 2H), 7.31–7.28 (m, 2H), 3.79 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 150.2, 149.9, 148.3, 131.8, 131.4, 129.2, 129.1 (q, JC-F = 33.0 Hz), 126.8, 121.7, 39.1; 19F NMR (470 MHz, CD3COCD3) δ − 58.5 (s); IR (KBr) υ 3060, 2925, 1680, 1608, 1545, 1500, 1346, 1263, 1110 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C12H8F3N3O4 315.0467; Found 315.0462.
5-(4-Chlorophenyl)-1-methyl-3-nitropyrazin-2(1H)-one (5c)
23.9 mg (45%); eluent (n-hexanes/acetone = 10:1 to 3:1); yellow solid; mp = 238.1–241.4 °C; 1H NMR (400 MHz, CD3COCD3) δ 8.78 (s, 1H), 7.86 (dt, J = 8.8, 2.8 Hz, 2H), 7.50 (dt, J = 8.4, 2.8 Hz, 2H), 3.81 (s, 3H); 13C NMR (100 MHz, CD3COCD3) δ 159.5, 135.5, 134.7, 133.8, 129.9, 128.3, 127.3, 105.0, 38.8; IR (KBr) υ 2925, 2856, 1678, 1606, 1543, 1489, 1340, 1267, 1196, 1086, 739 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C11H8ClN3O3 265.0254; Found 265.0250.
1-Methyl-3-nitro-5-(3-(trifluoromethyl)phenyl)pyrazin-2(1H)-one (5d)
34.2 mg (57%); eluent (n-hexanes/acetone = 10:1 to 3:1); yellow solid; mp = 159.2–161.2 °C; 1H NMR (500 MHz, CD3COCD3) δ 8.93 (s, 1H), 8.17–8.13 (m, 2H), 7.73 (dd, J = 3.5, 1.5 Hz, 2H), 3.83 (s, 3H); 13C NMR (125 MHz, CD3COCD3) δ 150.7, 149.3, 142.5, 136.2, 131.6 (q, JC-F = 31.2 Hz), 130.9, 129.4, 127.8, 126.3 (q, JC-F = 270.5 Hz), 125.7 (q, JC-F = 3.2 Hz), 122.2 (d, JC-F = 3.2 Hz), 38.9; 19F NMR (470 MHz, CD3COCD3) δ − 63.1 (s); IR (KBr) υ 3060, 2927, 1680, 1608, 1545, 1325, 1271, 1180, 1036 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C12H8F3N3O3 299.0518; Found 299.0515.
1-(Methoxymethyl)-3-nitro-5-phenylpyrazin-2(1H)-one (5e)
27.8 mg (53%); eluent (n-hexanes/acetone = 10:1 to 1:1); yellow oil; 1H NMR (500 MHz, CD3COCD3) δ 8.06 (s, 1H), 7.88–7.85 (m, 2H), 7.50–7.46 (m, 2H), 7.39 (tt, J = 7.0, 1.5 Hz, 1H), 5.54 (s, 2H), 3.52 (s, 3H); 13C NMR (125 MHz, CD3COCD3) δ 149.1, 134.8, 131.6, 130.1, 129.9, 129.5, 125.9, 124.4, 80.9, 58.3; IR (KBr) υ 2924, 2854, 1684, 1604, 1545, 1460, 1273 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C12H11N3O4 261.0750; Found 261.0747.
1-Benzyl-5-(2-methoxyphenyl)-3-nitropyrazin-2(1H)-one (5f)
30.4 mg (45%); eluent (n-hexanes/acetone = 10:1 to 1:1); yellow oil; 1H NMR (400 MHz, CD3COCD3) δ 8.20 (s, 1H), 7.62 (dd, J = 7.6, 1.6 Hz, 1H), 7.58 (ddd, J = 9.2, 7.6, 2.0 Hz, 1H), 7.43–7.41 (m, 2H), 7.35 (td, J = 7.2, 2.0 Hz, 2H), 7.27 (tt, J = 6.8, 1.6 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 7.07 (td, J = 7.6, 0.8 Hz, 1H), 4.54 (d, J = 6.0 Hz, 2H), 3.73 (s, 3H); 13C NMR (100 MHz, CD3COCD3) δ 192.7, 165.9, 160.7, 140.0, 135.5, 131.1, 129.2, 128.6, 127.9, 126.1, 121.5, 113.3, 56.4, 43.2; IR (KBr) υ 3240, 3074, 2584, 2318, 1683, 1645, 1580, 1485, 1462, 1439, 1310, 1250, 1207, 1165, 1117, 1022, 928 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C18H15N3O4 337.1063; Found 337.1062.
2-((3-Nitro-2-oxo-5-phenylpyrazin-1(2H)-yl)methyl)benzonitrile (5g)
40.6 mg (61%); eluent (n-hexanes/acetone = 10:1 to 3:1); yellow oil; 1H NMR (400 MHz, CD3COCD3) δ 8.90 (s, 1H), 7.88–7.85 (m, 3H), 7.73–7.67 (m, 2H), 7.58 (td, J = 7.6, 2.0 Hz, 1H), 7.51–7.47 (m, 2H), 7.50 (tt, J = 7.2, 1.6 Hz, 1H), 5.66 (s, 2H); 13C NMR (100 MHz, CD3COCD3) δ 153.1, 148.9, 138.5, 134.8, 134.5, 134.3, 134.1, 130.4, 130.3, 129.9, 129.8, 129.5, 125.9, 117.9, 112.9, 53.7; IR (KBr) υ 2956, 2924, 2225, 1668, 1593, 1525, 1448, 1344, 1213 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C18H12N4O3 332.0909; Found 332.0907.
1-(4-Methoxyphenyl)-3-nitro-5-phenylpyrazin-2(1H)-one (5h)
33.2 mg (51%); eluent (n-hexanes/acetone = 10:1 to 3:1); yellow solid; mp = 149.4–150.6 °C; 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H), 7.75–7.72 (m, 2H), 7.47–7.38 (m, 5H), 7.05 (dt, J = 8.8, 3.6 Hz, 2H), 3.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.5, 160.9, 147.9, 133.1, 131.1, 130.5, 130.4, 129.3, 127.0, 125.3, 121.6, 115.2, 55.9; IR (KBr) υ 3060, 1682, 1606, 1512, 1467, 1265 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C17H13N3O4 323.0906; Found 323.0906.
1-(4-Acetylphenyl)-3-nitro-5-phenylpyrazin-2(1H)-one (5i)
30.2 mg (45%); eluent (n-hexanes/acetone = 10:1 to 4:1); yellow oil; 1H NMR (500 MHz, CD3COCD3) δ 8.63 (s, 1H), 8.21 (dt, J = 9.0, 2.0 Hz, 2H), 7.91 (dq, J = 8.5, 1.0 Hz, 2H), 7.88 (dt, J = 8.5, 2.5 Hz, 2H), 7.50–7.46 (m, 2H), 7.40 (tt, J = 7.5, 1.0 Hz, 1H), 2.68 (s, 3H); 13C NMR (125 MHz, CD3COCD3) δ 197.2, 148.6, 143.2, 138.9, 134.7, 132.9, 130.2, 130.1, 129.8, 129.5, 127.8 (two carbons overlap), 126.0, 26.9; IR (KBr) υ 2924, 2854, 1685, 1599, 1545, 1360, 1265, 1194 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C18H13N3O4 335.0906; Found 335.0905.
3-Nitro-5-phenyl-1-(m-tolyl)pyrazin-2(1H)-one (5j)
31.4 mg (51%); eluent (n-hexanes/acetone = 10:1 to 4:1); yellow solid; mp = 141.3–144.1 °C; 1H NMR (400 MHz, CD3COCD3) δ 8.56 (s, 1H), 7.93–7.90 (m, 2H), 7.53–7.52 (m, 1H), 7.50–7.49 (m, 2H), 7.47–7.44 (m, 2H), 7.39 (tt, J = 7.2, 1.2 Hz, 2H), 2.44 (s, 3H); 13C NMR (100 MHz, CD3COCD3) δ 150.2, 148.7, 140.4, 139.9, 134.8, 133.4, 131.2, 130.1, 129.8, 129.4, 127.7, 125.9, 124.8, 124.3, 21.2; IR (KBr) υ 3059, 2925, 2854, 1684, 1603, 1545, 1489, 1454, 1342, 1269, 1194 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C17H13N3O3 307.0957; Found 307.0956.
General procedure for the gram scale experiment of 1a
To an oven-dried round bottom flask charged with 1-methylquinoxalin-2(1H)-one (1a) (1.0 g, 6.3 mmol, 100 mol %) was added t-butyl nitrite (2a) (2.25 mL, 18.9 mmol, 300 mol %) and CH3CN (60 mL) under O2 atmosphere at room temperature. After using O2 balloon, the reaction mixture was allowed to stir at 60 °C for 20 h. The reaction mixture was cooled to room temperature, diluted with EtOAc (25 mL) and concentrated in vacuo. The residue was purified by flash column chromatography (n-hexanes/EtOAc = 10:1 to 2:1) to afford 3a (0.69 g) in 68% yield.
General procedure for the gram scale experiment of 4a
To an oven-dried sealed tube charged with 1-methyl-5-phenylpyrazin-2(1H)-one (4a) (1.0 g, 5.4 mmol, 100 mol %) was added t-butyl nitrite (2a) (3.2 mL, 27.0 mmol, 500 mol %) and CH3CN (65 mL) under O2 atmosphere at room temperature. After using O2 balloon, the reaction mixture was allowed to stir at 60 °C for 28 h. The reaction mixture was cooled to room temperature, diluted with EtOAc (25 mL) and concentrated in vacuo. The residue was purified by flash column chromatography (n-hexane/acetone = 10:1 to 3:1) to afford 5a (0.81 g) in 65% yield.
General procedure and characterization data for the reduction of nitro group on 3a
To an oven-dried sealed tube charged with 1-methyl-7-nitroquinoxalin-2(1H)-one (3a) (41.0 mg, 0.2 mmol, 100 mol %), iron (46.9 mg, 0.84 mmol, 420 mol %), ammonium chloride powder (71.7 mg, 1.34 mmol, 670 mol %) were added MeOH/THF/H2O (1:1:1, 4.5 mL) at room temperature. The reaction mixture was allowed to stir in an oil bath for 12 h at 60 °C. The reaction mixture was cooled to room temperature, filtered through Celite, rinsing with methanol, and the volatiles were removed under reduced pressure. The aqueous residue was diluted with water, saturated NaHCO3 solution, and extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (n-hexanes/acetone = 10:1 to 2:1) to afford 6a (22.8 mg) in 65% yield.
7-Amino-1-methylquinoxalin-2(1H)-one (6a)
22.8 mg (65%); eluent (n-hexanes/acetone = 10:1 to 2:1); brown oil; 1H NMR (400 MHz, DMSO-d6) δ 7.74 (s, 1H), 7.44 (d, J = 8.4 Hz, 1H), 6.60 (dd, J = 8.8, 2.4 Hz, 1H), 6.47 (d, J = 2.4 Hz, 1H), 6.13 (brs, 2H), 3.46 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 155.0, 152.1, 141.3, 135.4, 130.9, 125.2, 111.3, 95.6, 28.2; IR (KBr) υ 3367, 3197, 2924, 2854, 1732, 1604, 1535, 1462, 1379, 1342, 1267 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C9H9N3O 175.0746; Found 175.0744.
General procedure and characterization data for the thiocarbonylation of 3a into 6b
To an oven-dried sealed tube charged with 1-methyl-7-nitroquinoxalin-2(1H)-one (3a) (41.0 mg, 0.2 mmol, 100 mol %) and Lawesson’s reagent (242.7 mg, 0.6 mmol, 300 mol %) was added toluene (2 mL) under air at room temperature. The reaction mixture was allowed to stir in an oil bath for 12 h at 120 °C. The reaction mixture was cooled to room temperature, diluted with EtOAc (5 mL) and concentrated in vacuo. The aqueous residue was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (n-hexanes/acetone = 10:1 to 1:2) to afford 6b (25.8 mg) in 58% yield.
1-Methyl-7-nitroquinoxaline-2(1H)-thione (6b)
25.8 mg (58%); eluent (n-hexanes/acetone = 10:1 to 1:2); orange oil; 1H NMR (400 MHz, CD3COCD3) δ 8.79 (s, 1H), 8.60 (d, J = 2.4 Hz, 1H), 8.27 (dd, J = 8.8, 2.4 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 4.30 (s, 3H); 13C NMR (100 MHz, CD3COCD3) δ 179.7, 160.1, 140.9, 132.3 (two carbons overlap), 120.5, 112.4 (two carbons overlap), 37.7; IR (KBr) υ 3060, 2924, 2854, 1684, 1550, 1516, 1460, 1350, 1267, 1103 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C9H7N3O2S 221.0259; Found 221.0256.
General procedures for the control experiment using a radical scavenger TEMPO
To an oven-dried sealed tube charged with 1-methylquinoxalin-2(1H)-one (1a) (32.0 mg, 0.2 mmol, 100 mol %) and TEMPO (312.5 mg, 2.0 mmol, 10.0 equiv.) were added t-butyl nitrite (2a) (71.4 µL, 0.6 mmol, 300 mol %) and CH3CN (2 mL) under O2 atmosphere at room temperature. After using O2 balloon, the reaction mixture was allowed to stir in an oil bath for 8 h at 60 °C. The reaction mixture was cooled to room temperature, diluted with EtOAc (4 mL) and concentrated in vacuo. On TLC the desired product 3a was not detected.
General procedure and characterization data for the reaction of 1a and 2a with radical polymerization mediator 1,1-diphenylethylene (2b)
To an oven-dried sealed tube charged with 1-methylquinoxalin-2(1H)-one (1a) (32.0 mg, 0.2 mmol, 100 mol %) and 1,1-diphenylethylene (2b) (72.1 mg, 0.4 mmol, 200 mol %) were added t-butyl nitrite (2a) (71.4 µL, 0.6 mmol, 300 mol %) and CH3CN (2 mL) under O2 atmosphere at room temperature. After using O2 balloon, the reaction mixture was allowed to stir in an oil bath for 8 h at 60 °C. The reaction mixture was cooled to room temperature, diluted with EtOAc (5 mL) and concentrated in vacuo. The aqueous residue was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (n-hexanes/acetone = 10:1 to 4:1) to afford 7a (25.2 mg) in 56% yield.
(2-Nitroethene-1,1-diyl)dibenzene (7a)
25.2 mg (56%); eluent (n-hexanes/acetone = 10:1 to 4:1); yellow oil; 1H NMR (400 MHz, CD3COCD3) δ 7.72 (s, 1H), 7.51–7.42 (m, 6H), 7.40–7.37 (m, 2H), 7.27–7.24 (m, 2H); 13C NMR (100 MHz, CD3COCD3) δ 149.9, 137.9, 136.9, 136.0, 131.6, 129.8, 129.7, 129.6, 129.5, 129.3; IR (KBr) υ 3059, 2925, 2854, 1610, 1574, 1510, 1495, 1444, 1308, 1267 cm−1; HRMS (quadrupole, EI) m/z: [M]+ Calcd for C14H11NO2 225.0790; Found 225.0791.
Results
Our optimization was performed by investigating the coupling reaction of 1-methylquinoxalin-2(1H)-one (1a) with tert-butyl nitrite (2a), as shown in Table 1.

The nitration reaction of 1a was initiated by using tert-butyl nitrite (2a) to deliver C7-nitrated quinoxalinone 3a in 12% yield, and no formation of other regioisomers including C5-nitrated adduct 3aa was observed (Table 1, entry 1). The chemical structure of C7-nitrated quinoxalinone 3a (CCDC 2099185) was elucidated by the X-ray crystallographic analysis (Fig. 3).

X-ray crystallographic data of 3a
Solvent screening revealed that this coupling reaction displayed the increased reactivity in CH3CN solvent to give 3a in 40% yield (Table 1, entries 2 − 4). Addition of oxidants such as K2S2O8, Na2S2O8, and AgNO2 were found to be unsatisfactory in this transformation (Table 1, entries 5 − 7). To our delight, this reaction smoothly proceeded with three equiv. of 2a to afford the desired product 3a in 76% yield along with C5-nitrated compound 3aa in 9% yield (Table 1, entry 8). The reaction temperature is quite pivotal for this transformation, as shown in entries 9 and 10. It should be noted that molecular oxygen was needed for the formation of both 3a and 3aa (Table 1, entry 11), revealing that a NO radical, derived from the decomposition of tert-butyl nitrite, could be readily oxidized into a reactive NO2 radical by molecular oxygen (O2 gas). Finally, when the reaction was performed with increased loading of 2a, the lower formation of our desired product was observed (Table 1, entries 12 and 13).
With the optimal reaction conditions in hand, the scope of quinoxalinones was examined as shown in Table 2. The linear and branched N-alkylated quinoxalinones 1b − 1e were found to be suitable substrates for this coupling reaction to afford C7-nitrated quinoxalinones 3b − 3e in high yields. It is noteworthy that a linear alkyl halide 1e was completely compatible under the current reaction conditions, and the tolerance of bromo moiety presents valuable opportunities for further versatile synthetic transformations. In addition, N-benzylated quinoxalinones 1f and 1g were also coupled with 2a to provide the corresponding products 3f (55%) and 3g (31%). To our pleasure, the current protocol could be applied to N-arylated quinoxalinones 1h − 1k, producing the desired products 3h − 3k without undergoing the C−H nitration on the N-aryl ring. It is mentioned that electron-deficient NO2 (1i) and CO2Et (1k) groups on the N-aryl moiety were found to be comparatively less reactive in this transformation, presumably due to the destabilization of radical and carbocation intermediates. To observe the steric and electronic effects on the quinoxalinone framework, the reactions of 1l with 2a under the standard and modified reaction conditions were subjected to afford the C7-nitrated adduct 3l in 20% and 35% yields. It should be mentioned that the nitration reaction of C7-substituted quinoxalinone 1m preferentially occurred at the C5-position, affording the nitrated product 3m as a single regioisomer in 45% yield.
With successfully screening results of quinoxalinone substrates, the substrate scope of various 5-aryl pyrazin-2-ones 4a − 4j was evaluated, as shown in Table 3. The reaction of 5-phenyl pyrazinone 4a with 2a under the modified reaction conditions (5 equiv. of 2a, 28 h) provided 5a in 72% yield. Additionally, N-alkyl-5-aryl-substituted pyrazinones 4b–4e reacted with 2a to afford C3-nitrated pyrazinone adducts 5b–5e in moderate to good yields. In addition, N-benzyl-substituted pyrazinones 4f and 4g were also compatible under the current reaction conditions to give the corresponding products 5f (45%) and 5g (61%). The complete regioselectivity was observed in all cases. Finally, the C3-nitration reaction of N-aryl-substituted pyrazinones 4h–4j smoothly proceeded, resulting in the formation of the desired products 5h–5j. The functional group compatibility of nitrile and acetyl moieties (5g and 5i) allows further synthetic elaboration of the products.
To demonstrate the robustness and practicality of this process, the scale-up experiments and synthetic transformations were performed (Fig. 4). The nitration reaction of 1a was readily scaled up to 1 g (6.3 mmol) for the formation of 3a (0.69 g) in 68% yield. In addition, the gram-scale reaction of 4a (1 g, 5.4 mmol) with 2a was successfully achieved to afford 0.81 g of 5a in 65% yield. Meanwhile, the selective reduction of a nitro moiety of the product 3a was performed by the single electron reduction protocol using by Fe/NH4Cl to furnish the desired aniline adduct 6a in 65% yield. Moreover, treatment of 3a with Lawesson’s reagent resulted in the formation of 7-nitroquinoxaline-2(1H)-thione 6b in 58% yield.

Scale-up experiments and synthetic transformations
Discussion
To support the mechanistic pathway for this reaction, the nitration reaction of 1a and 2a was performed in the presence of a radical scavenger TEMPO (Fig. 5). No formation of C7-nitrated quinoxalinone 3a was observed. The nitration reaction was completely inhibited by 1,1-diphenylethylene (2b) as a radical polymerization mediator, and 1,1-diphenyl-2-nitroethylene (7a) was obtained in 56% yield. These results support that a radical pathway is involved in this process.

Mechanistic investigations
Based on preliminary mechanistic investigation and reported literatures (Liang et al. 2015; Chun et al. 2018), a proposed reaction mechanism is outlined in Fig. 6. In the presence of molecular oxygen, a reactive NO2 radical can be derived from the thermal decomposition of tBuONO, followed by subsequent aerobic oxidation of a NO radical. A NO2 radical can undergo the radical addition into the C7-position on quinoxalinone 1a, affording intermediate A. The single-electron transfer (SET) process by the assistance of NO2 or tBuO radical followed by aromatization provides the C7-nitrated product 3a. In case of the nitration of pyrazinone 4a, the radical addition can occur at the C3-position, delivering a nitrogen radical species C, which further undergoes the SET reaction and elimination reaction to produce 6a. The site-selectivity between the C7- and C3-positions of quinoxalinone 1a can be rationalized by the electronic density between the electron-rich aromatic ring and electron-deficient N-heterocycle ring in the electrophilic radical addition step. Moreover, the C7-selectivity over C6,C8-positions on aromatic ring of 1a can be explained by the relative stability of a radical intermediate A. However, the site-selectivity between C7- and C5-position still remains unclear, and the detailed mechanistic investigations on the site-selectivity of this process are underway.

Proposed reaction mechanism for the site-selective C–H nitration
In summary, we described the synthesis of biologically relevant C7-nitrated quinoxalinones and C3-nitrated pyrazinones through metal-free C–H nitration with t-butyl nitrite. From the mechanistic point of view, the radical addition to quinoxalinones with t-butyl nitrite exclusively occurred at the electron-rich aromatic region beyond electron-deficient N-heterocycle ring. In contrast, the nitration reaction of pyrazinones readily takes place at the C3-position via the single electron transfer process of a nitrogen radical intermediate followed by elimination reaction. This protocol is characterized by the scale-up compatibility, mild reaction conditions, and excellent functional group tolerance. The selective reduction of a NO2 group and thiocarbonylation on the synthesized products highlight the importance of the developed methodology.
Change history
13 November 2021
A Correction to this paper has been published: https://doi.org/10.1007/s12272-021-01362-2
References
An W, Choi SB, Kim N, Kwon NY, Ghosh P, Han SH, Mishra NK, Han S, Hong S, Kim IS (2020) C2-selective C–H methylation of heterocyclic N-oxides with sulfonium ylides. Org Lett 22:9004–9009. https://doi.org/10.1021/acs.orglett.0c03403
Chun R, Kim S, Han SH, Pandey AK, Mishra NK, Kim IS (2018) Site-selective C–H nitration of N-aryl-7-azaindoles under palladium(II) catalysis. Tetrahedron Lett 59:3848–3852. https://doi.org/10.1016/j.tetlet.2018.09.025
Fan FR, Yao Y, Cai L, Cheng L, Tour JM, Bard AJ (2004) Structure-dependent charge transport and storage in self-assembled monolayers of compounds of interest in molecular electronics: effects of tip material, headgroup, and surface concentration. J Am Chem Soc 126:4035–4042. https://doi.org/10.1021/ja0359815
Fan Z, Ni J, Zhang A (2016) Meta-selective CAr-H nitration of arenes through a Ru3(CO)12-catalyzed ortho-metalation strategy. J Am Chem Soc 138:8470–8475. https://doi.org/10.1021/jacs.6b03402
Fors BP, Buchwald SL (2009) Pd-catalyzed conversion of aryl chlorides, triflates, and nonaflates to nitroaromatics. J Am Chem Soc 131:12898–12899. https://doi.org/10.1021/ja905768k
Freeman CD, Klutman NE, Lamp KC (1997) Metronidazole. a therapeutic review and update. Drugs 54:679–708. https://doi.org/10.2165/00003495-199754050-00003
Ghosh P, Kwon NY, Han S, Kim S, Han SH, Mishra NK, Jung YH, Chung SJ, Kim IS (2019) Site-selective C–H alkylation of diazine N-oxides enabled by phosphonium ylides. Org Lett 21:6488–6493. https://doi.org/10.1021/acs.orglett.9b02365
Ghosh P, Kwon NY, Kim S, Han S, Lee SH, An W, Mishra NK, Han SB, Kim IS (2021) C–H methylation of iminoamido heterocycles with sulfur ylides. Angew Chem Int Ed 60:191–196. https://doi.org/10.1002/anie.202010958
Guo J, Zhang L, Du X, Zhang L, Cai Y, Xia Q (2021) Metal-free direct oxidative C−N bond coupling of quinoxalin-2(1H)-ones with azoles under mild conditions. Eur J Org Chem 2021:2230–2238. https://doi.org/10.1002/ejoc.202100269
Han S, Chakrasali P, Park J, Oh H, Kim S, Kim K, Pandey AK, Han SH, Han SB, Kim IS (2018) Reductive C2-alkylation of pyridine and quinoline N-oxides using Wittig reagents. Angew Chem Int Ed 57:12737–12740. https://doi.org/10.1002/anie.201807159
Katayev D, Pfister KF, Wendling T, Gooßen LJ (2014) Copper-mediated ortho-nitration of (hetero)arenecarboxylates. Chem Eur J 20:9902–9905. https://doi.org/10.1002/chem.201403363
Kilpatrick B, Heller M, Arns S (2013) Chemoselective nitration of aromatic sulfonamides with tert-butyl nitrite. Chem Commun 49:514–516. https://doi.org/10.1039/C2CC37481A
Koley D, Colón OC, Savinov SN (2009) Chemoselective nitration of phenols with tert-butyl nitrite in solution and on solid support. Org Lett 11:4172–4175. https://doi.org/10.1021/ol901731w
Lee H, Kang D, Han SH, Chun R, Pandey AK, Mishra NK, Hong S, Kim IS (2019) Allylic acetals as acrolein oxonium precursors in tandem C–H allylation and [3+2] dipolar cycloaddition. Angew Chem Int Ed 58:9470–9474. https://doi.org/10.1002/anie.201903983
Li YM, Wei XH, Li XA, Yang SD (2013) Metal-free carbonitration of alkenes using K2S2O8. Chem Commun 49:11701–11703. https://doi.org/10.1039/C3CC47287F
Li YX, Li LH, Yang YF, Hua HL, Yan XB, Zhao LB, Zhang JB, Ji FJ, Liang YM (2014) Direct oxidative nitration of aromatic sulfonamides under mild conditions. Chem Commun 50:9936–9938. https://doi.org/10.1039/C4CC03784G
Liang YF, Li X, Wang X, Yan Y, Feng P, Jiao N (2015) Aerobic oxidation of PdII to PdIV by active radical reactants: direct C–H nitration and acylation of arenes via oxygenation process with molecular oxygen. ACS Catal 5:1956–1963. https://doi.org/10.1021/cs502126n
Liu YK, Lou SJ, Xu DQ, Xu ZY (2010) Regiospecific synthesis of nitroarenes by palladium-catalyzed nitrogen-donor-directed aromatic C–H nitration. Chem Eur J 16:13590–13593. https://doi.org/10.1002/chem.201002581
Majhi B, Kundu D, Ahammed S, Ranu BC (2014) tert-Butyl nitrite mediated regiospecific nitration of (E)-azoarenes through palladium-catalyzed directed C–H activation. Chem Eur J 20:9862–9866. https://doi.org/10.1002/chem.201403325
Manna S, Maity S, Rana S, Agasti S, Maiti D (2012) ipso-Nitration of arylboronic acids with bismuth nitrate and perdisulfate. Org Lett 14:1736–1739. https://doi.org/10.1021/ol300325t
McNamara YM, Cloonan SM, Knox AJ, Keating JJ, Butler SG, Peters GH, Meegan MJ, Williams DC (2011) Synthesis and serotonin transporter activity of 1,3-bis(aryl)-2-nitro-1-propenes as a new class of anticancer agents. Bioorg Med Chem 19:1328–1348. https://doi.org/10.1016/j.bmc.2010.11.054
Mishra NK, Sharma S, Park J, Han S, Kim IS (2017) Recent advances in catalytic C(sp2)–H allylation reactions. ACS Catal 7:2821–2847. https://doi.org/10.1021/acscatal.7b00159
Mishra NK, Park J, Oh H, Han SH, Kim IS (2018) Recent advances in N-heterocycles synthesis through catalytic C–H functionalization of azobenzenes. Tetrahedron 74:6769–6794. https://doi.org/10.1016/j.tet.2018.10.010
Nepali K, Lee HY, Liou JP (2019) Nitro-group-containing drugs. J Med Chem 62:2851–2893. https://doi.org/10.1021/acs.jmedchem.8b00147
Olah GA, Malhotra R, Narang, SC (1990) Nitration methods and mechanisms. Wiley-VCH, Weinheim. https://doi.org/10.1002/recl.19901091207
Ono N (2001) The nitro group in organic synthesis. Wiley-VCH, New York. https://doi.org/10.1002/0471224480
Pandey AK, Han SH, Mishra NK, Kang D, Lee SH, Chun R, Hong S, Park JS, Kim IS (2018) Synthesis of 2-benzazepines from benzylamines and MBH adducts under rhodium(III) catalysis via C(sp2)–H functionalization. ACS Catal 8:742–746. https://doi.org/10.1021/acscatal.7b03812
Park MS, Moon K, Oh H, Lee JY, Ghosh P, Kang JY, Park JS, Mishra NK, Kim IS (2021) Synthesis of (2H)-indazoles and dihydrocinnolinones through annulation of azobenzenes with vinylene carbonate under Rh(III) catalysis. Org Lett 23:5518–5522. https://doi.org/10.1021/acs.orglett.1c01866
Patel SS, Patel DB, Patel HD (2021) Synthetic protocols for aromatic nitration: a review. ChemistrySelect 6:1337–1356. https://doi.org/10.1002/slct.202004695
Rebstock MC, Crooks HM, Controulis J, Bartz QR (1949) Chloramphenicol (Chloromycetin).1 IV.1achemical studies. J Am Chem Soc 71:2458–2462. https://doi.org/10.1021/ja01175a065
Sambiagio C, Schönbauer D, Blieck R, Dao-Huy T, Pototschnig G, Schaaf P, Wiesinger T, Zia MF, Wencel-Delord J, Besset T, Maes BUW, Schnürch M (2018) A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem Soc Rev 47:6603–6743. https://doi.org/10.1039/C8CS00201K
Shen T, Yuan Y, Jiao N (2014) Metal-free nitro-carbocyclization of activated alkenes: a direct approach to synthesize oxindoles by cascade C–N and C–C bond formation. Chem Commun 50:554–556. https://doi.org/10.1039/C3CC47336H
Wan L, Qiao K, Yuan X, Zheng MW, Fan BB, Di ZC, Zhang D, Fang Z, Guo K (2017) Nickel-catalyzed regioselective C–H bond mono- and bis-nitration of aryloxazolines with tert-butyl nitrite as nitro source. Adv Synth Catal 359:2596–2604. https://doi.org/10.1002/adsc.201700186
Wu X, Neumann H, Beller M (2011a) Palladium-catalyzed Sonogashira reactions of aryl amines with alkynesvia in situ formation of arenediazonium salts. Chem Commun 47:7959–7961. https://doi.org/10.1039/C1CC12552D
Wu X, Schranck J, Neumann H, Beller M (2011b) Convenient and mild synthesis of nitroarenes by metal-free nitration of arylboronic acids. Chem Commun 47:12462–12463. https://doi.org/10.1039/C1CC15484B
Xie F, Qi Z, Li X (2013) Rhodium(III)-catalyzed azidation and nitration of arenes by C–H activation. Angew Chem Int Ed 52:11862–11866. https://doi.org/10.1002/anie.201305902
Zhang L, Liu Z, Li H, Fang G, Barry BD, Belay TA, Bi X, Liu Q (2011) Copper-mediated chelation-assisted ortho nitration of (hetero)arenes. Org Lett 13:6536–6539. https://doi.org/10.1021/ol2028288
Zolfigol MA, Khazaei A, Moosavi-Zare AR, Zare A, Kruger HG, Asgari Z, Khakyzadeh V, Kazem-Rostami M (2012) Design of ionic liquid 3-methyl-1-sulfonic acid imidazolium nitrate as reagent for the nitration of aromatic compounds by in situ generation of NO2 in acidic media. J Org Chem 77:3640–3645. https://doi.org/10.1021/jo300137w
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (Nos. 2019R1A4A2001451 and 2020R1A2C3005357).
Funding
This study was funded by National Research Foundation of Korea (Nos. 2019R1A4A2001451 and 2020R1A2C3005357).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts to declare relevant to this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to a retrospective Open Access order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moon, J., Ji, H.K., Ko, N. et al. Site-selective and metal-free C–H nitration of biologically relevant N-heterocycles. Arch. Pharm. Res. 44, 1012–1023 (2021). https://doi.org/10.1007/s12272-021-01351-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-021-01351-5