
Abstract
α-Synuclein oligomers and Ca2+ dyshomeostasis have been thoroughly investigated with respect to the pathogenesis of Lewy body disease (LBD). In LBD, α-synuclein oligomers exhibit a neuron-specific cytoplasmic distribution. Highly active neurons and neurons with a high Ca2+ burden are prone to damage in LBD. The neuronal vulnerability may be determined by transneuronal axonal transmission of the pathological processes; however, this hypothesis seems inconsistent with pathological findings that neurons anatomically connected to LBD-vulnerable neurons, such as neurons in the ventral tegmentum, are spared in LBD. This review focuses on and discusses the crucial roles played by α-synuclein oligomers and Ca2+ dyshomeostasis in early intraneural pathophysiology in LBD-vulnerable neurons. A challenging view is proposed on the synergy between retrograde transport of α-synuclein and vesicular Ca release, whereby neuronal vulnerability is propagated backward along repeatedly activated signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pathological hallmarks of Lewy body disease (LBD), [also known as Parkinson’s disease (PD) and dementia with Lewy bodies] are Lewy bodies and Lewy neurites, which are composed of insoluble α-synuclein fibrils (Irvine et al. 2008). The LBD pathological process targets specific subcortical and cortical neurons in addition to dopaminergic neurons in the substantia nigra pars compacta (SNc) (Braak et al. 2003). In the peripheral nervous system, LBD pathology mainly appears in the autonomic nervous system (Braak et al. 2003; Surmeier and Sulzer 2013; Kalia and Lang 2015). In the central nervous system, lesions initially occur in the dorsal motor nucleus of the vagus nerve (DMV) and the anterior olfactory nucleus (Braak et al. 2003; Surmeier and Sulzer 2013; Kalia and Lang 2015). In the brainstem, Lewy pathology and cell loss have been observed in the region of the DMV, the medullary reticular formation, the raphe nuclei, the locus coeruleus, the pedunculopontine nuclei, and the SNc (Braak et al. 2003; Surmeier and Sulzer 2013; Kalia and Lang 2015). The Lewy pathology in the brainstem and the anterior olfactory nucleus expands into related areas such as the nucleus basalis of Meynert, the amygdala, and the cerebral cortex (Braak et al. 2003; Surmeier and Sulzer 2013; Kalia and Lang 2015). This selective neuronal involvement is the background not only of classical parkinsonian motor symptoms, but also of heterogeneous non-motor features, including autonomic dysfunction, olfactory dysfunction, sleep disorders, psychiatric symptoms, and cognitive impairment, all of which lower the quality of life in LBD patients (Chaudhuri et al. 2006). In contrast, neurons in the ventral tegmental area (VTA) and globus pallidus, striatal cholinergic interneurons, and cerebellar Purkinje neurons, which are anatomically connected to PD-vulnerable neurons, are not involved in PD (Braak et al. 2003; Surmeier and Sulzer 2013). Given that selective neuronal vulnerability in slowly progressing LBD pathology is incompatible with the simple idea of transneuronal retrograde axonal spreading of LBD pathology along anatomical connectivities (Del Tredici and Braak 2020), what is the basis of selective neuronal fragility in LBD?
This review focuses on two key features of neurons in the LBD brain that contribute to the pathophysiology of LBD-vulnerable neurons: α-synuclein oligomers and calcium dyshomeostasis. Also discussed is the recent finding that α-synuclein oligomers trigger an aberrant form of robust-firing-induced calcium release from the endoplasmic reticulum (ER), which is mediated by direct binding with calcium binding protein 1 (CaBP1), a neuron-specific member of the calmodulin superfamily. This binding disrupts the normal Ca2+- and IP3-dependent regulation of inositol 1, 4, 5-trisphosphate (IP3) receptors (IP3Rs) (Yamamoto et al. 2019). The present review proposes that this signaling-path-specific calcium dysregulation uniquely contributes to early oligomeric α-synuclein-mediated pathophysiology in LBD-vulnerable neurons.
α-Synuclein oligomers: a pivotal factor in LBD pathogenesis
α-Synuclein is highly expressed in the brain and is enriched at presynaptic terminals. It augments assembly of the SNARE machinery, and plays a role in neurotransmitter release and protection of nerve terminals against injury (Kalia et al. 2013). The main physiological form of α-synuclein in the brain appears to be an unfolded monomer, but it also forms soluble oligomeric species that are toxic to neurons (Irvine et al. 2008; Dehay et al. 2015; Ingelsson 2016). The presence of α-synuclein oligomers has been demonstrated not only with recombinant proteins or in cell culture and in vivo models, but also in post-mortem brain tissue from LBD patients (Cappai et al. 2005; Yamakawa et al. 2010; Sharon et al. 2003; Paleologou et al. 2009). Proximity ligation assays of LBD brain tissue can detect α-synuclein oligomers, which can go undetected by conventional immunohistochemistry using anti-α-synuclein antibodies (Roberts et al. 2015). α-Synuclein oligomers can be observed not only in the neuropil, but also in the cytosol of LBD-vulnerable neurons, which can precede the development of classical PD lesions, such as pale bodies or Lewy bodies. (Roberts et al. 2015).
In sporadic PD, genetic risk factors, such as mutations in the glucocerebrosidase gene (GBA) (Oeda et al. 2015; Schapira 2015), and susceptibility variants in SNCA, MAPT, LRRK2, PARK16, and BST1 have been identified by genome-wide association studies (Satake et al. 2009; Simón-Sánchez et al. 2009). The aggregation of α-synuclein is upregulated in variants of α-synuclein (Irvine et al. 2008) and LRRK2 (Aasly et al. 2014). α-Synuclein BAC transgenic mice show oligomeric forms of α-synuclein in the regions that are specifically affected in LBD, including the olfactory bulb, cerebral cortex, striatum and SNc, and exhibit non-motor symptoms, such as rapid eye movement sleep behavior disorder-like behavior and hyposmia (Taguchi et al. 2020). Environmental risk factors, including exposure to MPTP and rotenone, or oxidative stress, also cause LBD pathology (Irvine et al. 2008), while exposure to metals and pesticides can increase α-synuclein aggregation (Uversky et al. 2002; Irvine et al. 2008). α-Synuclein oligomer toxicity occurs via several intracellular mechanisms, including mitochondrial dysfunction, ER stress, impaired autophagy-lysosomal pathways (Dehay et al. 2015; Ingelsson 2016), and neuroinflammation (Kim et al. 2013; Rocha et al. 2018). Furthermore, oligomeric and fibril species of α-synuclein can be transferred between neurons, thereby propagating α-synuclein pathologies (Hansen et al. 2011; Kim et al. 2019). This wide array of studies indicates that the α-synuclein oligomer is a key toxic molecule of LBD-vulnerable neurons before Lewy bodies are formed and neuronal loss occurs (Fig. 1).

α-Synuclein oligomers and intraneural calcium dyshomeostasis play key roles in the pathophysiological mechanism of LBD. Genetic and environmental risk factors promote LBD pathology, including the production of α-synuclein (αSn) oligomers and the disturbance of Ca2+ homeostasis in LBD-vulnerable neurons, mutually strengthening the metabolic and proteostatic burden, and increasing the neuron-to-neuron propagation of αSn pathology and neuroinflammation. These pathophysiological alterations cause selective neuronal vulnerability and the spreading of LBD pathology. The metabolic and proteostatic burden includes mitochondrial dysfunction, the autophagy and lysosomal pathway and the ubiquitin proteasome system (UPS), and ER stress. LBD-vulnerable neurons share the features underlying Ca2+ dyshomeostasis, such as being autonomous pacemaker neurons, having broad and slow spikes, and low levels of Ca2+ binding protein. Note that neurons that do not have such Ca2+-related properties, such as neocortical neurons, are also prone to damage in LBD patients with cognitive decline
Dysregulated Ca2+ homeostasis: a key feature of selective neuronal fragility in LBD
Dysregulation of Ca2+ homeostasis is a core pathological mechanism in LBD. It triggers the formation of α-synuclein oligomers, mitochondrial and ER stress, and inhibition of autophagy and lysosomal pathways, thereby leading to neurodegeneration (Surmeier and Sulzer 2013). In SNc neurons of aging mice, Ca2+ entry via CaV1.3, an L-type voltage-dependent Ca2+ channel (VDCC) during autonomous action potentials causes oxidative stress and cell damage (Chan et al. 2007; Surmeier and Sulzer 2013). Epidemiological studies indicate that L-type VDCC (L-VDCC) blockers diminish the risk of PD (Ritz et al. 2010; Ascherio and Schwarzschild 2016). Moreover, soluble oligomeric α-synuclein induces Ca2+ influx and seeding (Danzer et al. 2007), and promotes Ca2+-induced mitochondrial dysfunction (Luth et al. 2014). Taken together, these observations raise the possibility that the close relationship between α-synuclein oligomers and the dysregulation of intracellular Ca2+ handling by intraneural organelles may play a central role in LBD pathogenesis.
In general, LBD-vulnerable neurons, such as neurons in the SNc, the locus coeruleus, the raphe nuclei and the nucleus basalis of Meynert, have a common physiological phenotype; an autonomous pacemaker, broad and slow spiking, and low levels of Ca2+-binding protein expression (Surmeier and Sulzer 2013). These physiological properties lead to increased cytosolic Ca2+ and augment the metabolic burden in these neurons that is critical for selective neuronal degeneration (Surmeier and Sulzer 2013). Of note, the neurons in the VTA, which are LBD-resistant, have high neuronal activities as autonomous pacemakers, but express a high level of Ca2+-binding protein, indicating the importance of not only activity-dependent, but also signaling-path-specific Ca2+ burden for the selective neuronal vulnerability in LBD (Surmeier and Sulzer 2013). Isradipine, a dihydropyridine calcium-channel (CaV1.3) blocker approved for the treatment of hypertension, is neuroprotective in animal models of PD (Chan et al. 2007; Guzman et al. 2010; Ilijic et al. 2011). This neuroprotective effect is mediated by inhibition of plasma membrane L-type calcium channels, which trigger mitochondrial oxidant stress and turnover (Guzman et al. 2010; Ilijic et al. 2011). These studies prompted a clinical trial evaluating the efficacy of isradipine to slow the clinical progression of PD in previously untreated early-stage PD patients. However, this trial indicated no significant effect (The Parkinson Study Group STEADY-PD III Investigators 2020). Attention has, therefore, been directed to other Ca2+ channel-dependent mechanisms of activity-dependent Ca2+ dyshomeostasis and signaling-path-specific selective fragility of neurons in LBD brains.
Lewy pathology is neuron-specific in LBD, suggesting a crucial role of intraneural α-synuclein in LBD pathology that is distinct from the cytoplasmic α-synuclein inclusions of glial cells in multiple system atrophy (Jucker and Walker 2013; Del Tredici and Braak 2020). Lewy pathology appears in neocortical pyramidal neurons, which do not have properties related to Ca2+ dyshomeostasis, such as being autonomous pacemaker cells, exhibiting broad and slow spikes and low levels of Ca2+ binding protein (Yamamoto et al. 2019). This pathology contributes to dementia in LBD, the second major cause of degenerative dementia (Braak et al. 2003; Chaudhuri et al. 2006). An unsolved question is, therefore, how intraneural α-synuclein oligomers pathologically can alter neuronal activity and intracellular Ca2+ dynamics in neocortical neurons. By using intracellular injection of bioactive molecules or proteins such as inositol trisphosphate (IP3), homer1a and amyloid-β through a patch pipette, how Ca2+ and K+ channels are involved in the regulation or pathophysiological change of neocortical pyramidal cell excitability and Ca2+ dynamics are demonstrated (Sakagami et al. 2005; Yamamoto et al. 2000, 2002a, 2002b, 2005, 2011). Interestingly, the results obtained using these methods correspond to those observed in neurons with physiologically produced IP3 or homer1a proteins in the cytoplasm, or in neurons of 3xTg Alzheimer’s disease model mice (Cui et al. 2007; Nakamura et al. 1999; Sakagami et al. 2005; Stutzmann et al. 2003; Yamamoto et al. 2000, 2002a, 2002b, 2005, 2011). The same methodology was used to introduce α-synuclein protein into pyramidal neurons in cortical slices from mice to investigate the effects and mechanisms of intracellular α-synuclein oligomers on neuronal excitability and Ca2+ dynamics, as described below (Yamamoto et al. 2019).
Aberrant activity-dependent calcium release from IP3 receptors in central neurons caused by the association of α-synuclein oligomers with CABP1
To clarify pathophysiological changes in neuronal activity regulating intraneural Ca2+ induced by α-synuclein oligomers, whole-cell recordings were obtained from pyramidal neurons located in slices of the mouse frontal cortex (Yamamoto et al. 2019). Immunoblotting analysis using anti-α-synuclein antibodies revealed that the recombinant α-synuclein incubated with dopamine at 37 °C for 3 days produced higher-order oligomers (αSNo) and fibrils, while the recombinant α-synuclein incubated without dopamine for 3 days was free of higher-order oligomers (αSN) (Yamamoto et al. 2019). After filtering to remove α-synuclein fibrils, αSNo and αSN solutions were used as pipette solutions, and the properties of action potentials in current clamp mode and the current charges during spike afterhyperpolarization (AHP) in voltage clamp mode were examined (Yamamoto et al. 2019). This examination revealed that intracellular application of αSNo significantly reduced spike frequency during current injection, elongated the duration of spike AHP, and enlarged AHP current charge compared with αSN (Yamamoto et al. 2019; Fig. 2). This αSNo-mediated alteration was triggered by spike-induced Ca2+ release from IP3Rs functionally coupled with L-VDCC and small conductance Ca2+ activated K+ (SK) channels under the application of blockers for the channels or receptors responsible for intraneural Ca2+ dynamics (Yamamoto et al. 2019; Fig. 2). This Ca2+-dependent functional triad consisting of L-VDCCs, IP3Rs and SK channels is well established and is linked to spike-triggered Ca2+ inflow and Ca2+ release (Ca2+-induced Ca2+ release; CICR) from IP3Rs in neurons of the neocortex and amygdala, and contributes to the regulation of neuronal excitability and synaptic plasticity Yamamoto et al. 2000, 2002a, b, Faber et al. 2010, Power and Sah 2005, 2008; Yamada et al. 2004; Fig. 2c, iii). In contrast with previous reports that emphasized how the physiological upregulation of IP3 turnover is finely tuned by synaptic stimulation or neuromodulation and is necessary for spike-induced or IP3-induced Ca2+ release from IP3Rs in central neurons (Cui et al. 2007; Faber et al. 2010, Nakamura et al. 1999; Power and Sah 2005, 2008; Stutzmann et al. 2003; Yamada et al. 2004; Yamamoto et al. 2000, 2002a, 2002b; Fig. 2c, ii), the oligomeric α-synuclein-mediated CICR from IP3Rs presented here was independent of increased IP3 production, because the phospholipase C (PLC) blocker, U73122, failed to inhibit it (Yamamoto et al. 2019; Fig. 2b). Such an unusual mode of CICR provoked by highly frequent neuronal activity, independent of IP3 turnover, does not usually occur in central neurons because the regulation of IP3R gating exhibits bell-shaped dependence on somatic Ca2+ concentration (Bezprozvanny et al. 1991). This mode of CICR can therefore be regarded as pathological, forcing an excess Ca2+ burden on neurons, surpassing a negative feedback regulation of spike firing (Fig. 2c, iii). Accordingly, via this channel coupling, α-synuclein oligomers provoke the aberrant CICR from IP3Rs, which is triggered by Ca2+ influx via L-VDCCs during repetitive firing, followed by elongation of SK channel opening (the prolongation of IAHP) and decreased spike frequency (Yamamoto et al. 2019). Consequently, in neocortical pyramidal neurons, the occurrence of this aberrant mode of CICR can be detected by examining the elevation of IAHP charge and the reduction in spike frequency (Fig. 2c i, iii; Yamamoto et al. 2019).

The electrophysiological detection and the mechanism of oligomeric α-synuclein-mediated aberrant Ca2+-induced Ca2+ release (CICR). a Aberrant CICR was detected by a decrease in spike frequency (SK) and an enhancement of IAHP (afterhyperpolarization current) in αSNo (including higher-order α-synuclein oligomers)-applied neurons compared with αSN (free from higher-order α-synuclein oligomers)-applied neurons. After filtering to remove α-synuclein fibrils, αSNo and αSN solutions were used as pipette solutions for current clamp and voltage clamp recordings (Yamamoto et al. 2019). b The mechanism of oligomeric α-synuclein-mediated aberrant CICR was examined using drugs to modulate the channels and receptors that control intraneural Ca2+ dynamics, and biomolecules or antibodies that regulate these channels and receptors (Yamamoto et al. 2019). The aberrant CICR is blocked by the extracellular application (non-italic text) of blockers of L-VDCC, SK, and IP3R, a Ca2+ chelator, and an ER store depletor, and by the intracellular application (italic text) of calcium binding protein 1 (CaBP1), which is a neuron-specific regulator of IP3R gating and directly binds higher order α-synuclein oligomers (Yamamoto et al. 2019). The intracellular application of IP3 and CaBP1 antibodies (Ab) both occlude the effect of αSNo. c A scheme illustrating the aberrant CICR during burst firing (from Yamamoto et al. 2019 with partial modification). (i) Endogenous CaBP1 (blue circle) binds IP3Rs and maintains Ca2+-dependent inactivation of IP3Rs (white rectangle). (ii) IP3 elevation controlled by finely tuned neurotransmission or neuromodulation is required for physiological CICR from IP3Rs. GPCR/Gq: G-protein-coupled receptor/Gq protein. (iii) Intraneural α-synuclein oligomer (red ellipse) binds to endogenous CaBP1 and allows aberrant CICR from IP3Rs (red rectangle) independent of IP3 turnover, which enhances Ca2+ dysregulation. (iv) CaBP1 Ab binds endogenous CaBP1, prevents CaBP1 regulation of IP3Rs and triggers aberrant CICR in a similar manner to the α-synuclein oligomer. (v) CaBP1 binds α-synuclein oligomer and blocks oligomeric α-synuclein-mediated aberrant CICR
IP3R has two separate binding sites for Ca2+ and IP3, which are allosterically regulated by the two ligands. The binding of one ligand facilitates additional binding of the other (Berridge 1998; Verkhratsky 2005); therefore, IP3R responds to the increase in neuronal cytosolic Ca2+ and IP3, opens, and releases Ca2+ from the ER in an activity-dependent manner (Yamamoto et al. 2000, 2002a, b; Yamada et al. 2004, Nakamura et al. 1999, Larkum et al. 2003). The PLC blocker, U73122, failed to block Ca2+ release but intracellular application of IP3 occluded the effect of αSNo; therefore, α-synuclein oligomers modulate IP3R gating and mediate the aberrant form of CICR from IP3Rs during repetitive spikes, without enhancing Ca2+ influx or IP3 production in neocortical neurons (Yamamoto et al. 2019; Fig. 2c, iii).
The gating of IP3R is not only regulated by IP3 or Ca2+ binding, but also by various proteins that interact with IP3R (Choe and Ehrlich 2006; Foskett et al. 2007); therefore, αSNo could be linked to a protein that directly binds IP3R and regulates IP3R gating in central neurons. Among the binding partners of IP3R, Ca2+-binding protein 1 (CaBP1) is the most promising candidate, because CaBP1 is (1) a Ca2+-binding protein expressed in the cytosol of rodent and human central neurons (Laube et al. 2002; Bernstein et al. 2003; Kim et al. 2014), (2) a protein that preferentially interacts with oligomeric α-synuclein (Betzer et al. 2015), and (3) a binding partner and negative regulator of IP3Rs under high intraneural Ca2+ concentration by means of Ca2+-dependent inactivation (Haynes et al. 2004; Kasri et al. 2004; Li et al. 2013). CaBP1 is a neuronal Ca2+-binding protein splice variant, a sub-branch of the calmodulin superfamily, which includes Ca2+-sensor proteins that regulate various Ca2+ channel targets (Yang et al. 2002; Haeseleer et al. 2000). CaPB1 has four EF-hand Ca2+-binding motifs, and can bind and regulate IP3R under high intraneural Ca2+ concentration (Haynes et al. 2004; Kasri et al. 2004; Li et al. 2013). Once αSNo binds to CaBP1 and pulls it away from IP3R, thus disrupting Ca2+-dependent regulation of IP3R, an aberrant CICR from IP3R can occur without enhancing Ca2+ influx or cytosolic IP3 concentration.
The effects of intracellular application of a CaBP1 antibody (Ab) and CaBP1 on αSNo-mediated change were compatible with this hypothesis. CaBP1 Ab occluded the effect of αSNo, which was blocked by nifedipine and heparin, indicating that αSNo-mediated capture of CaBP1 is necessary and sufficient for the aberrant CICR from IP3R (Yamamoto et al. 2019; Fig. 2c, iv, v). By contrast, calmodulin, which is another binding partner and regulator of IP3R (Yang et al. 2002; Taylor and Tovey 2010) and directly binds α-synuclein (Lee et al. 2002; Martinez et al. 2003), and a calmodulin antibody, failed to counteract αSNo-mediated action (Yamamoto et al. 2019; Fig. 2b). These electrophysiological observations were further supported by immunoprecipitation experiments that demonstrated the direct binding of higher-order α-synuclein oligomers larger than 100 kDa with CaBP1 (Yamamoto et al. 2019). Together, these electrophysiological findings demonstrate that the target of α-synuclein oligomers was CaBP1, and confirmed the direct association of α-synuclein oligomers greater than 100 kDa with CaBP1, resulting in aberrant CICR from IP3R (Yamamoto et al. 2019). These results are consistent with previous studies showing that transgenic α-synuclein mice exhibit augmented long-lasting Ca2+ transients in response to repetitive stimulation in vivo (Reznichenko et al. 2012), and that neocortical pyramidal cell excitability is reduced by injecting α-synuclein oligomers (Kaufmann et al. 2016), but emphasize that α-synuclein oligomers cause this activity-dependent signaling-path-specific CICR surpassing the regulation of spike firing.
IP3 and CaBP1 have opposing effects on the IP3R channel under high intracellular Ca2+ concentrations (Haynes et al. 2004; Karsi et al. 2004; Li et al. 2013). IP3 obstructs the inter-subunit interaction of IP3R and encourages IP3R channel opening (Choe and Ehrlich 2006; Li et al. 2013). By contrast, CaBP1 binds IP3R and prevents the inter-subunit interaction of IP3R when the somatic concentration of IP3 is low, thereby hindering IP3R channel opening in a Ca2+-dependent manner Choe and Ehrlich 2006; Li et al. 2013; Fig. 2c i). These mechanisms clarify why IP3 and CaBP1 Ab occlude and heparin and CaBP1 inhibit the action of αSNo (Fig. 2c, ii, iv, v); oligomeric α-synuclein-mediated removal of CaBP1-mediated regulation of IP3R, but not the Ca2+ buffering effect of CaBP1, is essential for aberrant CICR from IP3R (Fig. 2c, iii).
A potential mechanism of oligomeric α-synuclein-mediated calcium dysregulation in early pathology of LBD-vulnerable neurons
IP3R is localized on ER membrane connected with mitochondria, known as the mitochondria-associated ER membrane, and increased Ca2+ transfer from the ER via IP3Rs to mitochondria inhibits autophagy (Szabadkai et al., 2008; Decuypere et al. 2011). Chronic aberrant CICR from the IP3R in the presence of intraneural α-synuclein oligomers may increase the risk of activity-dependent mitochondrial stress through the IP3R-mitochondrial connection, and may lead to neuronal fragility in oligomeric α-synuclein-bearing neurons. This notion is supported by previous studies indicating that α-synuclein can alter mitochondrial Ca2+ homeostasis by enhancing ER-mitochondria interactions (Cali et al. 2012), and that α-synuclein can be localized to the mitochondria-associated ER membrane (Guardia-Laguarta et al. 2014).
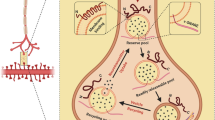
The ER is a continuous “neuron-within-neuron” network that extends to all parts of the neuron, including the spines, cell soma, and synaptic endings, and that supports regional and long-distance Ca2+ homeostasis (Berridge 1998; Öztürk et al. 2020). Ca2+ signals can transmit through the cytosol by CICR from the ER, and induce regional or global communication within the cell, at a pace that is slower than action potential propagation in the plasma membrane. CICR can be mediated by IP3Rs or ryanodine receptors and can be augmented by elevated cytosolic Ca2+ (Berridge 1998; Öztürk et al. 2020). The CICR from IP3Rs propagates as a Ca2+ wave along the ER via IP3Rs and ryanodine receptors throughout the somatodendritic portion and the nucleus (Power and Sah 2002; Watanabe et al. 2006; Ross 2012). This normal Ca2+ wave propagating from dendrite to soma and nucleus, is initiated only when strong synaptic stimulation or synaptic stimulation concurrent with repetitive spikes occurs (Power and Sah 2002; Watanabe et al. 2006; Ross 2012). This is mainly dependent on IP3R which is distributed in the soma and dendrites, thereby regulating neuronal excitability and synaptic plasticity (Power and Sah 2002; Watanabe et al. 2006; Ross 2012). In contrast, it is noteworthy that repetitive spikes alone, independent of synaptic input, are enough to cause the aberrant CICR from the ER via IP3Rs in the presence of α-synuclein oligomers (Yamamoto et al. 2019; Fig. 2c, iii). Subsequent to backpropagating action potentials from the soma to dendrites (Waters et al. 2005), this aberrant CICR can cause the chronic aberrant Ca2+ wave to be propagated along the soma-dendritic region (Yamamoto et al. 2019; Fig. 3). A sporadic PD risk gene, BST1, encodes cyclic ADP-ribose hydrolase 2, which synthesizes cyclic ADP-ribose, a ryanodine receptor agonist (Satake et al. 2009; Saad et al. 2011). BST1 variants can disturb normal channel function of the ryanodine receptor, another Ca2+ release channel in the ER, and can enhance the aberrant CICR via IP3Rs to promote propagation of dysregulated Ca2+ waves (Yamamoto et al. 2019; Fig. 3). Chronic occurrence of this aberrant CICR and Ca2+ wave may increase the risk of distinct activity-dependent Ca2+ dyshomeostasis and may lead to pathological intraneural spreading and neuronal fragility specific to repeatedly activated signaling path in oligomeric α-synuclein-bearing neurons, although this remains to be studied. Considering that oligomeric α-synuclein accumulates first at presynaptic terminals in LBD (Bridi et al. 2018), and that the pathological change of LBD-vulnerable neurons is a dying back-phenomenon, which initiates at synaptic terminals and progresses along the axon, affecting homeostasis and survival of neuronal cell bodies (Cheng et al. 2010; Bridi et al. 2018), a tentative mechanism of oligomeric α-synuclein-mediated aberrant calcium dyshomeostasis promoting early neuronal pathophysiological change in LBD-vulnerable neurons, is proposed in Fig. 3.

A potential mechanism of oligomeric α-synuclein-mediated calcium dyshomeostasis promoting early pathology in LBD-vulnerable neurons. a In LBD-vulnerable neurons, various LBD risk factors trigger a dying-back pathology that is initiated at synaptic terminals and progresses along the axon with oligomeric α-synuclein (αSn) appearing in cell bodies. During burst firing, the oligomeric αSn-mediated CICR from IP3Rs propagates as an aberrant Ca2+ wave via IP3Rs and ryanodine receptors along the somatodendritic portion of the ER (Ca2+: black dot). b A proposed retrograde bottom-up model of αSn propagation reinforced by the aberrant Ca2+ wave in LBD-vulnerable neurons. Retrograde propagation of αSn pathology (upper left) brings somatodendritic αSn oligomers, which cause chronic aberrant CICR propagation (aberrant Ca2+ wave) during burst firing (upper right). This Ca2+ dysregulation in turn promotes the Ca2+ burden, the secretion of αSn from dendrites (lower right), and transmission of αSn pathology to nearby synaptic terminals (lower left). Together with neuronal degeneration of LBD-vulnerable neurons, the retrograde propagation of αSn pathology in presynaptic LBD-vulnerable neurons is initiated (lower left, upper right). This mechanism specific to repeatedly activated signaling path can account for the propagation of LBD pathology
Immunohistochemical studies reveal that among central neurons, the expression level of CaBP1 is lowest in SNc neurons, which are the most fragile in PD (Laube et al. 2002; Kim et al. 2014). The scarcity of CaBP1 in SNc neurons suggests that the smallest amount of α-synuclein oligomers can induce the aberrant CICR described here, and that this may account for by far the highest vulnerability of SNc neurons. The L-VDCC blocker, isradipine, suppresses Ca2+ influx via CaV1.3 L-VDCC and inhibits degeneration of SNc neurons (Chan et al. 2007), although this drug failed to slow the progression of PD (The Parkinson Study Group STEADY-PD III Investigators 2020). Given that L-VDCC blockers suppress aberrant CICR from IP3Rs (Yamamoto et al. 2019), and lower the risk of PD (Ritz et al. 2010; Ascherio and Schwarzschild 2016), they may reduce the risk of aberrant CICR propagation along the ER in the somatodendritic area, and may have potential in protecting LBD-vulnerable neurons from damage and the spread of Lewy body pathology.
References
Aasly JO, Johansen KK, Brønstad G, Warø BJ, Majbour NK, Varghese S, Alzahmi F, Paleologou KE, Amer DA, Al-Hayani A, El-Agnaf OM (2014) Elevated levels of cerebrospinal fluid α-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front Aging Neurosci 6:248. https://doi.org/10.3389/fnagi.2014.00248
Ascherio A, Schwarzschild MA (2016) The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol 15:1257–1272. https://doi.org/10.1016/s1474-4422(16)30230-7
Bernstein HG, Seidenbecher CI, Smalla KH, Gundelfinger ED, Bogerts B, Kreutz MR (2003) Distribution and cellular localization of caldendrin immunoreactivity in adult human forebrain. J Histochem Cytochem 51:1109–1112. https://doi.org/10.1177/002215540305100816
Berridge MJ (1998) Neuronal calcium signaling. Neuron 21:13–26. https://doi.org/10.1016/s0896-6273(00)80510-3
Betzer C, Movius AJ, Shi M, Gai WP, Zhang J, Jensen PH (2015) Identification of synaptosomal proteins binding to monomeric and oligomeric alpha-synuclein. PLoS One 10:e0116473. https://doi.org/10.1371/journal.pone.0116473
Bezprozvanny I, Watras J, Ehrlich BE (1991) Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351:751–754. https://doi.org/10.1038/351751a0
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Bridi JC, Hirth F (2018) Mechanisms of α-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front Neurosci 12:80. https://doi.org/10.3389/fnins.2018.00080
Cali T, Ottolini D, Negro A, Brini M (2012) alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem 287:17914–17929. https://doi.org/10.1074/jbc.M111.302794
Cappai R, Leck SL, Tew DJ, Williamson NA, Smith DP, Galatis D, Sharples RA, Curtain CC, Ali FE, Cherny RA, Culvenor JG, Bottomley SP, Masters CL, Barnham KJ, Hill AF (2005) Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J 19:1377–1379. https://doi.org/10.1096/fj.04-3437fje
Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ (2007) ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447:1081–1086. https://doi.org/10.1038/nature05865
Chaudhuri KR, Healy DG, Schapira AHV (2006) Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol 5:235–245. https://doi.org/10.1016/S1474-4422(06)70373-8
Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67:715–725. https://doi.org/10.1002/ana.21995
Choe CU, Ehrlich BE (2006) The inositol 1,4,5-trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Sci STKE 363:re15. https://doi.org/10.1126/stke.3632006re15
Cui G, Bernier BE, Harnett MT, Morikawa H (2007) Differential regulation of action potential- and metabotropic glutamate receptor-induced Ca2+ signals by inositol 1,4,5-trisphosphate in dopaminergic neurons. J Neurosci 27:4776–4785. https://doi.org/10.1523/JNEUROSCI.0139-07.2007
Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M (2007) Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci 27:9220–9232. https://doi.org/10.1523/JNEUROSCI.2617-07.2007
Decuypere JP, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB (2011) The IP3 receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta 1813:1003–1013. https://doi.org/10.1016/j.bbamcr.2010.11.023
Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, Singleton A, Olanow CW, Merchant KM, Bezard E, Petsko GA, Meissner WG (2015) Targeting alpha-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol 14:855–866. https://doi.org/10.1016/S1474-4422(15)00006-X
Del Tredici K, Braak H (2020) To stage, or not to stage. Curr Opin Neurobiol 61:10–22. https://doi.org/10.1016/j.conb.2019.11.008
Faber ES (2010) Functional interplay between NMDA receptors, SK channels and voltage-gated Ca2+ channels regulates synaptic excitability in the medial prefrontal cortex. J Physiol 588:1281–1292. https://doi.org/10.1113/jphysiol.2009.185645
Foskett JK, White C, Cheung KH, Mak DO (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87:593–658. https://doi.org/10.1152/physrev.00035.2006
Guardia-Laguarta C. Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, Voos W, Schon EA, Przedborski S (2014) alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci 34:249–259. https://doi.org/10.1523/JNEUROSCI.2507-13.2014
Guzman JN, Sanchez-Padilla J, Wokosin D et al (2010) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468:696–700. https://doi.org/10.1038/nature09536
Haeseleer F, Sokal I, Verlinde CL, Erdjument-Bromage H, Tempst P, Pronin AN, Benovic JL, Fariss RN, Palczewski K (2000) Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J Biol Chem 275:1247–1260. https://doi.org/10.1074/jbc.275.2.1247
Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121:715–725. https://doi.org/10.1172/JCI43366
Haynes LP, Tepikin AV, Burgoyne RD (2004) Calcium-binding protein 1 is an inhibitor of agonist-evoked, inositol 1,4,5-trisphosphate-mediated calcium signaling. J Biol Chem 279:547–555. https://doi.org/10.1074/jbc.M309617200
Ilijic E, Guzman JN, Surmeier DJ (2011) The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol Dis 43:364–371. https://doi.org/10.1016/j.nbd.2011.04.007
Ingelsson M (2016) Alpha-synuclein oligomers-neurotoxic molecules in Parkinson’s disease and other Lewy body disorders. Front Neurosci 10:408. https://doi.org/10.3389/fnins.2016.00408
Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM (2008) Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol Med 14:451–464. https://doi.org/10.2119/2007-00100.Irvine
Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501:45–51. https://doi.org/10.1038/nature12481
Kalia LV, Kalia SK, McLean PJ, Lozano AM, Lang AE (2013) α-Synuclein oligomers and clinical implications for Parkinson disease. Ann Neurol 73:155–169. https://doi.org/10.1002/ana.23746
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet 386:896–912. https://doi.org/10.1016/S0140-6736(14)61393-3
Kasri NN, Holmes AM, Bultynck G, Parys JB, Bootman MD, Rietdorf K, Missiaen L, McDonald F, De Smedt H, Conway SJ, Holmes AB, Berridge MJ, Roderick HL (2004) Regulation of InsP3 receptor activity by neuronal Ca2+-binding proteins. Embo j 23:312–321. https://doi.org/10.1038/sj.emboj.7600037
Kaufmann TJ, Harrison PM, Richardson MJ, Pinheiro TJ, Wall MJ (2016) Intracellular soluble alpha-synuclein oligomers reduce pyramidal cell excitability. J Physiol 594:2751–2772. https://doi.org/10.1113/JP271968
Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ (2013) Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4:1562. https://doi.org/10.1038/ncomms2534
Kim KY, Scholl ES, Liu X, Shepherd A, Haeseleer F, Lee A (2014) Localization and expression of CaBP1/caldendrin in the mouse brain. Neuroscience 268:33–47. https://doi.org/10.1016/j.neuroscience.2014.02.052
Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S, Lee JH, Kim WR, Kook M, Foss CA, Shen C, Lee H, Kulkarni S, Pasricha PJ, Lee G, Pomper MG, Dawson VL, Dawson TM, Ko HS (2019) Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 103:627–641.e7. https://doi.org/10.1016/j.neuron.2019.05.035
Larkum ME, Watanabe S, Nakamura T, Lasser-Ross N, Ross WN (2003) Synaptically activated Ca2+ waves in layer 2/3 and layer 5 rat neocortical pyramidal neurons. J Physiol 549:471–488. https://doi.org/10.1113/jphysiol.2002.037614
Laube G, Seidenbecher CI, Richter K, Dieterich DC, Hoffmann B, Landwehr M, Smalla KH, Winter C, Bockers TM, Wolf G, Gundelfinger ED, Kreutz MR (2002) The neuron-specific Ca2+-binding protein caldendrin: gene structure, splice isoforms, and expression in the rat central nervous system. Mol Cell Neurosci 19:459–475. https://doi.org/10.1006/mcne.2001.1078
Lee D, Lee SY, Lee EN, Chang CS, Paik SR (2002) alpha-Synuclein exhibits competitive interaction between calmodulin and synthetic membranes. J Neurochem 82:1007–1017. https://doi.org/10.1046/j.1471-4159.2002.01024.x
Li C, Enomoto M, Rossi AM, Seo MD, Rahman T, Stathopulos PB, Taylor CW, Ikura M, Ames JB (2013) CaBP1, a neuronal Ca2+ sensor protein, inhibits inositol trisphosphate receptors by clamping intersubunit interactions. Proc Natl Acad Sci USA 110:8507–8512. https://doi.org/10.1073/pnas.1220847110
Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ (2014) Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem 289:21490–21507. https://doi.org/10.1074/jbc.M113.545749
Martinez J, Moeller I, Erdjument-Bromage H, Tempst P, Lauring B (2003) Parkinson’s disease-associated alpha-synuclein is a calmodulin substrate. J Biol Chem 278:17379–17387. https://doi.org/10.1074/jbc.M209020200
Nakamura T, Barbara J-G, Nakamura K, Ross WN (1999) Synerigistic release of Ca from InsP3-sensitive stores evoked by synaptic activation of mGlu receptors paired with backpropagating action potentials. Neuron 24:727–737. https://doi.org/10.1016/s0896-6273(00)81125-3
Oeda T, Umemura A, Mori Y, Tomita S, Kohsaka M, Park K, Inoue K, Fujimura H, Hasegawa H, Sugiyama H, Sawada H (2015) Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol Aging 36:3306–3313. https://doi.org/10.1016/j.neurobiolaging.2015.08.027
Öztürk Z, O’Kane CJ, Pérez-Moreno JJ (2020) Axonal Endoplasmic Reticulum Dynamics and Its Roles in Neurodegeneration. Front Neurosci 14:48. https://doi.org/10.3389/fnins.2020.00048
Paleologou KE, Kragh CL, Mann DM, Salem SA, Al-Shami R, Allsop D, Hassan AH, Jensen PH, El-Agnaf OM (2009) Detection of elevated levels of soluble alpha-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain 132:1093–1101. https://doi.org/10.1093/brain/awn349
Parkinson Study Group STEADY-PD III Investigators (2020) Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann Intern Med 172:591–598. https://doi.org/10.7326/M19-2534
Power JM, Sah P (2002) Nuclear calcium signaling evoked by cholinergic stimulation in hippocampal CA1 pyramidal neurons. J Neurosci 22:3454–3462. https://doi.org/10.1523/JNEUROSCI.22-09-03454.2002
Power JM, Sah P (2005) Intracellular calcium store filling by an L-type calcium current in the basolateral amygdala at subthreshold membrane potentials. J Physiol (Lond) 562:439–453. https://doi.org/10.1113/jphysiol.2004.076711
Power JM, Sah P (2008) Competition between calcium-activated K + channels determines cholinergic action on firing properties of basolateral amygdala projection neurons. J Neurosci 28:3209–3220. https://doi.org/10.1523/JNEUROSCI.4310-07.2008
Reznichenko L, Cheng Q, Nizar K, Gratiy SL, Saisan PA, Rockenstein EM, Gonzalez T, Patrick C, Spencer B, Desplats P, Dale AM, Devor A, Masliah E (2012) In vivo alterations in calcium buffering capacity in transgenic mouse model of synucleinopathy. J Neurosci 32:9992–9998. https://doi.org/10.1523/JNEUROSCI.1270-12.2012
Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S (2010) L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol 67:600–606. https://doi.org/10.1002/ana.21937
Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2015) Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 138:1642–1657. https://doi.org/10.1093/brain/awv040
Rocha EM, De Miranda B, Sanders LH (2018) Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis 109:249–257. https://doi.org/10.1016/j.nbd.2017.04.004
Ross WN (2012) Understanding calcium waves and sparks in central neurons. Nat Rev Neurosci 13:157–168. https://doi.org/10.1038/nrn3168
Saad M, Lesage S, Saint-Pierre A, Corvol JC, Zelenika D, Lambert JC, Vidailhet M, Mellick GD, Lohmann E, Durif F, Pollak P, Damier P, Tison F, Silburn PA, Tzourio C, Forlani S, Loriot MA, Giroud M, Helmer C, Portet F, Amouyel P, Lathrop M, Elbaz A, Durr A, Martinez M, Brice A; French Parkinson’s Disease Genetics Study Group. (2011) Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum Mol Genet 20:615–627 https://doi.org/10.1093/hmg/ddq497
Sakagami Y, Yamamoto K, Sugiura S, Inokuchi K, Hayashi T, Kato N (2005) Essential roles of Homer-1a in homeostatic regulation of pyramidal cell excitability: a possible link to clinical benefits of electroconvulsive shock. Eur J Neurosci 21:3229–3239. https://doi.org/10.1111/j.1460-9568.2005.04165.x
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41:1303–1307. https://doi.org/10.1038/ng.485
Schapira AH (2015) Glucocerebrosidase and Parkinson disease: recent advances. Mol Cell Neurosci 66:37–42. https://doi.org/10.1016/j.mcn.2015.03.013
Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ (2003) The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 37:583–595. https://doi.org/10.1016/s0896-6273(03)00024-2
Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T (2009 Dec) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41(12):1308–1312. https://doi.org/10.1038/ng.487
Stutzmann GE, LaFerla FM, Parker I (2003) Ca2+ signaling in mouse cortical neurons studied by two-photon imaging and photoreleased inositol triphosphate. J Neurosci 23:758–765. https://doi.org/10.1523/JNEUROSCI.23-03-00758.2003
Surmeier DJ, Sulzer D (2013) The pathology roadmap in Parkinson disease. Prion 7:85–91. https://doi.org/10.4161/pri.23582
Szabadkai G, Duchen MR (2008) Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 23:84–94. https://doi.org/10.1152/physiol.00046.2007
Taguchi T, Ikuno M, Hondo M, Parajuli LK, Taguchi K, Ueda J, Sawamura M, Okuda S, Nakanishi E, Hara J, Uemura N, Hatanaka Y, Ayaki T, Matsuzawa S, Tanaka M, El-Agnaf OMA, Koike M, Yanagisawa M, Uemura MT, Yamakado H, Takahashi R (2020) α-Synuclein BAC transgenic mice exhibit RBD-like behaviour and hyposmia: a prodromal Parkinson’s disease model. Brain 143:249–265. https://doi.org/10.1093/brain/awz380
Taylor CW, Tovey SC (2010) IP3 receptors: toward understanding their activation. Cold Spring Harb Perspect Biol 2:a004010. https://doi.org/10.1101/cshperspect.a004010
Uversky VN, Li J, Bower K, Fink AL (2002) Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: implications for Parkinson’s disease. Neurotoxicology 23:527–536. https://doi.org/10.1016/s0161-813x(02)00067-0
Verkhratsky A (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 85:201–279. https://doi.org/10.1152/physrev.00004.2004
Watanabe S, Hon M, Lasser-Ross N, Ross WN (2006) Modulation of calcium wave propagation in the dendrites and to the soma of rat hippocampal pyramidal neurons. J Physiol 575:455–468. https://doi.org/10.1113/jphysiol.2006.114231
Waters J, Schaefer A, Sakmann B (2005) Backpropagating action potentials in neurones: measurement, mechanisms and potential functions. Prog Biophys Mol Biol 87:145–170. https://doi.org/10.1016/j.pbiomolbio.2004.06.009
Yamada S, Takechi H, Kanchiku I, Kita T, Kato N (2004) Small-conductance Ca2+-dependent K+ channels are the target of spike-induced Ca2+ release in a feedback regulation of pyramidal cell excitability. J Neurophysiol 91:2322–2329. https://doi.org/10.1152/jn.01049.2003
Yamakawa K, Izumi Y, Takeuchi H, Yamamoto N, Kume T, Akaike A, Takahashi R, Shimohama S, Sawada H (2010) Dopamine facilitates alpha-synuclein oligomerization in human neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun 391:129–134. https://doi.org/10.1016/j.bbrc.2009.11.015
Yamamoto K, Hashimoto K, Isomura Y, Shimohama S, Kato N (2000) An IP3-assisted form of Ca2+-induced Ca2+ release in neocortical neurons. Neuroreport 11:535–539. https://doi.org/10.1097/00001756-200002280-00022
Yamamoto K, Hashimoto K, Nakano M, Shimohama S, Kato N (2002a) A distinct form of calcium release down-regulates membrane excitability in neocortical pyramidal cells. Neuroscience 109:665–676. https://doi.org/10.1016/s0306-4522(01)00486-9
Yamamoto K, Nakano M, Hashimoto K, Shimohama S, Kato N (2002b) Emergence of a functional coupling between inositol-1,4,5-trisphosphate receptors and calcium channels in developing neocortical neurons. Neuroscience 109:677–685. https://doi.org/10.1016/s0306-4522(01)00449-3
Yamamoto K, Sakagami Y, Sugiura S, Inokuchi K, Shimohama S, Kato N (2005) Homer 1a enhances spike-induced calcium influx via L-type calcium channels in neocortex pyramidal cells. Eur J Neurosci 22:1338–1348. https://doi.org/10.1111/j.1460-9568.2005.04278.x
Yamamoto K, Ueta Y, Wang L, Yamamoto R, Inoue N, Inokuchi K, Aiba A, Yonekura H, Kato N (2011) Suppression of a neocortical potassium channel activity by intracellular amyloid-beta and its rescue with Homer1a. J Neurosci 31:11100–11109. https://doi.org/10.1523/JNEUROSCI.6752-10.2011
Yamamoto K, Izumi Y, Arifuku M, Kume T, Sawada H (2019) α-Synuclein oligomers mediate the aberrant form of spike-induced calcium release from IP3 receptor. Sci Rep 9:15977. https://doi.org/10.1038/s41598-019-52135-3
Yang J, McBride S, Mak DO, Vardi N, Palczewski K, Haeseleer F, Foskett JK (2002) Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc Natl Acad Sci USA 99:7711–7716. https://doi.org/10.1073/pnas.102006299
Acknowledgements
This work was supported by a Grant-in-aid for scientific research (KAKENHI 23591265, KAKENHI 26461287, KAKENHI 20K07753) from the Japan Society for Promotion of Sciences. I express our gratitude to Dr. N. Kato (Kanazawa Medical University) and Dr. H. Sawada (NHO Utano National Hospital) for advice. I also thank Jeremy Allen, PhD, from Edanz Group (https://en-author-services.edanz.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamamoto, K. Complicity of α-synuclein oligomer and calcium dyshomeostasis in selective neuronal vulnerability in Lewy body disease. Arch. Pharm. Res. 44, 564–573 (2021). https://doi.org/10.1007/s12272-021-01334-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-021-01334-6