
Abstract
Seven new dianthrone glycosides, named polygonumnolides A1–B3 (1–7), were isolated from the 70 % EtOH extract of the dried roots of Polygonum multiflorum Thunb. using column chromatography and preparative high-performance liquid chromatography. Their structures were determined by 1D and 2D NMR and mass spectroscopy. The isolated compounds were evaluated for their cytotoxic effects against KB tumor cell lines and compounds 1–4 showed moderate cytotoxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The roots of Polygonum multiflorum Thunb. are used as a traditional Chinese medicinal herb to treat many diseases. They are applied as remedies for preventing hair loss and premature graying, strengthening bones and muscles, and treating seminal emission and menstrual and menopausal complaints (The State Pharmacopoeia Commission 2015). Chemical extractions of the roots of P. multiflorum resulted in the isolation of approximately 100 compounds (Lin et al. 2015), including anthraquinones, stilbenes, phenolic acid, phospholipids and flavones. In our continuing search for bioactive compounds, 70 % Et OH extract of the dried roots of P. multiflorum was investigated. Seven new dianthrone glycosides, named polygonumnolides A1–B3 (1–7), were isolated, and the structural elucidation of these new compounds are described herein as well as their cytotoxic effects against KB tumor cell lines.
Materials and methods
General experimental procedures
Optical rotations were acquired on a Jasco P-2000 polarimeter (Jasco Inc., Tokyo, Japan). UV data were recorded using a Jasco V-650 spectrophotometer (Jasco Inc., Tokyo, Japan). Experimental electronic circular dichroism spectra (ECD) were recorded on a Chirascan spectrophotometer. IR spectra were measured on a Nicolet iN 10 Micro FTIR spectrophotometer (Thermo Nicolet Inc., Waltham, MA, USA). NMR spectra were recorded on Varian Inova-300, 500 and 600 spectrophotometers (Varian Inc., Palo Alto, CA, USA). HRESI-MS were obtained using an Agilent 1100 UPLC-Q-TOF mass spectrometer (Agilent Technologies Ltd., Santa Clara, CA, USA). Column chromatography was performed with silica gel (200–300 mesh; Qingdao Marine Chemistry Company, Qingdao, China), Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) and reversed-phase C18 silica gel (40–60 µm, Alltech, Deerfield, IL, USA). Preparative high-performance liquid chromatography (HPLC) separations were carried out using a Shimadzu LC-10 A system equipped with a YMC-Pack ODS-A column (250 × 20 mm, 5 µm, Kyoto, Japan) and a Shimadzu SPD-20 A detector (Shimadzu).
Plant material
The dried roots of P. multiflorum Thunb. were collected from Deqing, Guangdong Province, People’s Republic of China, in October 2012, and identified by associate Prof. Ji Zhang (Research and Inspection Center of Traditional Chinese Medicine and Ethnomedicine, National Institutes for Food and Drug Control, State Food and Drug Administration). A voucher specimen (No. 060104) has been deposited at the Research and Inspection Center of Traditional Chinese Medicine and Ethnomedicine, National Institutes for Food and Drug Control, State Food and Drug Administration, Beijing 100050, People’s Republic of China.
Extraction and isolation
The roots of P. multiflorum Thunb. (28.0 kg) were extracted three times with 70 % EtOH under reflux and filtered. The filtrate was evaporated under reduced pressure at 50 °C to afford a crude extract (4.0 kg). The crude extract was partitioned with CH2Cl2 and H2O. The H2O fraction (3.5 kg) was loaded onto macroporous resin (DM-8) and eluted with a gradient of water and 95 % EtOH mixture (H2O, 25 % EtOH, 40 % EtOH, 55 % EtOH and 95 % EtOH) to give five fractions (A–E; fraction A: 2.0 kg; fraction B: 62.0 g; fraction C: 200.0 g; fraction D: 38.0 g; fraction E: 55.0 g). Fraction E was separated on a Sephadex LH-20 column using step gradient elution of methanol–water (from 60 to 100 % v/v) to give five fractions (A1–A5) based on RP-TLC analysis. Fraction A3 was further chromatographed on a RP-18 silica gel column using a step gradient elution of methanol–water (from 40 to 100 % v/v) to give 5 fractions (B1–B5) based on RP-TLC analysis.
Fraction B4 was purified on a Sephadex LH-20 column (100 % MeOH) to give fractions C4–C5 and isolated 2* (6.5 mg) and 4* (6.8 mg). Fraction C4 (50 mg) was further separated and purified by preparative HPLC (CH3CN/H2O, 50:50; YMC, 250 × 20 nm, S-5 µm; 210 nm; 5.0 mL/min) to yield 3* (7.0 mg, 54.0 min). Fraction C5 (50.0 mg) was separated and purified by preparative HPLC (CH3CN/H2O, 60:40; YMC, 250 × 20 nm, S-5 µm; 210 nm; 5.0 mL/min) to yield 1* (7.0 mg, 28.0 min). Fraction B2 was separated and purified by preparative HPLC (CH3CN/H2O, 35:65; YMC, 250 × 20 nm, S-5 µm; 210 nm; 5.0 mL/min) to yield 5* (7.0 mg, 31.0 min), 6* (6.0 mg, 36.0 min) and 7* (6.0 mg, 60.0 min), respectively.
Polygonumnolide A1 (1)
Yellow powder; [α] 25D −152° (c = 0.1, MeOH); UV (MeOH) λmax (logε): 207 (4.87), 280 (4.27), 351 (4.31) nm; ECD (c = 1.61 × 10−4 M, MeOH): Δε 392.0 nm −15.55, Δε 338.0 nm +20.35, Δε 279.0 nm −16.75; IR (KBr) νmax: 3361, 2919, 1620, 1599, 1489, 1376, 1333, 1254, 1176, 1160, 1074, 908, 862, 788 cm−1; 1H NMR (CD3OD, 500 MHz) and 13C NMR (CD3OD, 125 MHz) data, see Tables 1 and 2; HRESI–MS: m/z 685.1940 [M–H]− (calcd for C37H34O13, 685.1921).
Polygonumnolide A2 (2)
Yellow powder; [α] 25D −27.3° (c = 0.11, MeOH); UV (MeOH) λmax (logε): 207 (4.88), 279 (4.26), 351 (4.31) nm; ECD (c = 1.33 × 10−4 M, MeOH): Δε 387.0 nm +5.54, Δε 341.5 nm −5.12, Δε 317.0.0 nm +4.12, Δε 295.0 nm −5.76, Δε 278.0 nm +3.88; IR (KBr) νmax: 3404, 2924, 1619, 1599, 1488, 1378, 1334, 1256, 1218, 1177, 1074, 906, 861, 799 cm−1; 1H NMR (CD3COCD3, 600 MHz) and 13C NMR (CD3COCD3, 150 MHz) data, see Tables 1 and 2; HRESI-MS: m/z 685.1906 [M–H]− (calcd for C37H34O13, 685.1921).
Polygonumnolide A3 (3)
Yellow powder; [α] 25D −220° (c = 0.10, MeOH); UV (MeOH) λmax (logε): 207 (4.86), 279 (4.26), 351 (4.30) nm; ECD (c = 1.33 × 10−4 M, MeOH): Δε 366.5 nm +9.17, Δε 316.0.5 nm −14.47, Δε 288.0 nm +0.81, Δε 276.0 nm −2.28; IR (KBr) νmax: 3391, 2921, 1619, 1598, 1489, 1369, 1332, 1252, 1177, 1160, 1072, 1036, 907, 860, 788 cm−1; 1H NMR (CD3COCD3, 500 MHz) and 13C NMR (CD3COCD3, 125 MHz) data, see Tables 1 and 2; HRESI-MS: m/z 685.1943 [M–H]− (calcd for C37H34O13, 685.1921).
Polygonumnolide A4 (4)
Yellow powder; [α] 25D −250° (c = 0.06, MeOH); UV (MeOH) λmax (logε): 207 (4.86), 279 (4.25), 351 (4.29) nm; ECD (c = 1.33 × 10−4 M, MeOH): Δε 390.0 nm −14.66, Δε 343.0.5 nm +17.65, Δε 305.0 nm −17.56, Δε 280.0 nm +8.44, Δε 267.5 nm +7.77; IR (KBr) νmax: 3375, 2918, 1619, 1600, 1489, 1372, 1331, 1252, 1176, 1160, 1073, 1036, 907, 861, 789 cm−1; 1H NMR (CD3OD, 500 MHz) and 13C NMR (CD3OD, 125 MHz) data, see Tables 1 and 2; HRESI-MS: m/z 685.1898 [M–H]− (calcd for C37H34O13, 685.1921).
Polygonumnolide B1 (5)
Yellow powder; [α] 25D −120° (c = 0.10, MeOH); UV (MeOH) λmax (logε): 208(4.81), 279 (4.41), 348 (4.38) nm; ECD (c = 1.95 × 10−4 M, MeOH): Δε 382.0 nm −13.84, Δε 319.0 nm +28.56, Δε 279.5 nm −16.53; IR (KBr) νmax: 3381, 2922, 1630, 1599, 1493, 1354, 1334, 1259, 1217, 1178, 1075, 906, 865, 793 cm−1; 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz) data, see Tables 3 and 4; HRESI-MS: m/z 847.2453 [M–H]− (calcd for C43H44O18, 847.2454).
Polygonumnolide B2 (6)
Yellow powder; [α] 25D −110° (c = 0.10, MeOH); UV (MeOH) λmax (logε): 208(4.81), 279 (4.41), 346 (4.38) nm; ECD (c = 1.15 × 10−4 M, MeOH): Δε 367.5 nm +3.45, Δε 300.0 nm −5.96; IR (KBr) νmax: 3382, 2918, 1630, 1599, 1492, 1353, 1334, 1295, 1217, 1177, 1076, 905, 864, 790 cm−1; 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz) data, see Tables 3 and 4; HRESI-MS: m/z 847.2455 [M–H]− (calcd for C43H44O18, 847.2454).
Polygonumnolide B3 (7)
Yellow powder; [α] 25D −230° (c = 0.10, MeOH); UV (MeOH) λmax (logε): 208 (4.81), 279 (4.41), 346 (4.38) nm; ECD (c = 1.30 × 10−4 M, MeOH): Δε 376.0 nm +23.08, Δε 316.5 nm −47.53, Δε 279.0 nm +19.51; IR (KBr) νmax: 3382, 2918, 1630, 1599, 1492, 1353, 1334, 1295, 1217, 1177, 1076, 905, 864, 790 cm−1; 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz) data, see Tables 3 and 4; HRESI-MS: m/z 847.2452 [M–H]− (calcd for C43H44O18, 847.2454).
Enzyme hydrolysis of compounds
Compounds 1–7 (3.0 mg each) (Tian et al. 2013) were enzymatically hydrolyzed by β-glucosidase (10.0 mg) from almonds (CAS 9001-22-3) at 30 °C for 12 h, respectively. The reaction mixtures were extracted with EtOAC (3 × 10 mL). The aqueous layers were frozen for 12 h in order to remove organic solvent and freeze-dried to obtain the monosaccharides of 1–7. l-Cysteine methyl ester hydrochloride (1.5 mg) was added to solutions of the monosaccharides of compounds 1–7 and d-glucose in pyridine (3.0 mL) and kept at 60 °C for 1 h. The reaction mixtures were cooled in an ice-water bath, trimethylsilylimidazole (1.0 mL) was added and the mixtures heated to 60 °C for 30.0 min. The reaction mixtures were partitioned between H2O (2 mL) and n-hexane (3 × 10 mL). The n-hexane extracts of each digest were subjected to GC analysis, run on a Agilent 7890A gas chromatograph equipped with a Agilent HP-5 capillary column (60.0 m × 0.32 mm × 1.0 µm) and an H2 flame ionization detector with the following conditions: column temperature, 160–280 °C; ramp, 5 °C/min and maintained at 280 °C for 20.0 min; carrier gas, N2 (1 mL/min); injector and detector temperature, 300 °C; injection volume, 5.0 µL; and split ratio, 1/30. The configuration of the monosaccharides in each sample was determined by comparing the retention time of the derivatives with that of an authentic sample. All the samples gave a single peak with the same retention time of 40.6 min and carbohydrates in 1–7 were determined to all be d-glucose.
Cytotoxicity assay
A tetrazolium-based colorimetric assay (methyl thiazolyl tetrazolium assay, MTT assay) was used to assess the cytotoxicity of 1–7 against KB tumor cell lines with taxol used as the positive control. The assays were performed according to a published technique (Zhang et al. 2001).
Results and discussion
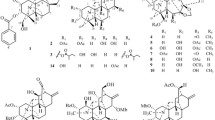
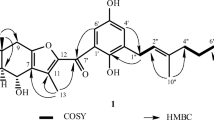
Compound 1 (Fig. 1) was obtained as a yellow powder. Its molecular formula, C37H34O13, was deduced from HRESI-MS from the peak at m/z 685.1940 [M–H]−, which corresponded to 21 indices of hydrogen deficiency. The IR spectrum showed strong absorption bands at 1620 and 1599 cm−1 that were assigned to carbonyl groups, a peak from chelated hydroxyl groups at 3361 cm−1 and a peak from aromatic ring functionalities at 1489 cm−1. The UV spectrum showed absorption maxima at 207, 280 and 351 nm, which were very close to those of previously reported dianthrone derivatives (Lemli et al. 1964; Vandenberg and Labadie 1981; Gizachew et al. 1993). The 1H NMR (Table 1) and 1H–1H COSY spectra (Fig. 2) displayed the signals of eight meta-coupled aromatic protons [δ H 6.45 (1H, s, H-2), 5.58 (1H, br s, H-4), 6.30 (1H, br s, H-5), 6.83 (1H, d, J = 2.0 Hz, H-7), 6.59 (1H, s, H-2′), 6.34 (1H, br s, H-4′), 5.45 (1H, br s, H-5′), and 6.18 (1H, d, J = 2.0 Hz, H-7′)], two methyl groups [δ H 2.07 (3H, s, Me-3) and 2.29 (3H, s, Me-3′)], one methoxy group at δ H 3.74 (3H, s, OCH 3 -6′), two vicinal methine protons [δ H 4.14 (1H, d, J = 3.5 Hz, H-10) and 4.08 (1H, d, J = 3.5 Hz, H-10′)], and one β-glucopyranosyl anomeric proton at δ H 4.60 (1H, d, J = 7.5 Hz, H-1′′).

Structures of compounds 1–7 from the roots of Polygonum multiflorum Thunb

Key 1H–1H COSY and HMBC correlations of compounds 1 and 5
Comparison of the 1D NMR spectrum of 1 with those of physcion dianthrones, emodin dianthrones, and physcion-emodin dianthrones (Donald et al. 1976; Du et al. 2008; Monache et al. 1991) suggested 1 could be a C-10/C-10′ isomer of physcion-emodin dianthrone glycosides. The 13C NMR and DEPT spectra (Table 2) exhibited 37 carbon signals, including 31 carbon signals of physcion-emodin dianthrones that were categorized by DEPT and HSQC techniques into 18 quaternary carbons, including with two carbonyl groups [δ C 191.3 (C-9′) and 188.5 (C-9)], 10 methine groups [δ C 117.3 (C-2), 121.8 (C-4), 112.5 (C-5), 107.7 (C-7), 117.7 (C-2′), 122.2 (C-4′), 109.7 (C-5′), 100.7 (C-7′), 57.7 (C-10) and 56.7 (C-10′)], two methyl groups [δ C 21.8 (Me-3) and 22.1 (Me-3′)], one methoxy group δ C 56.5 (OMe-6′)] and six carbon signals characteristic of glucose [δ C 106.1, 74.9, 78.8, 71.2, 77.4, 62.7].
The HMBC spectrum (Fig. 2) of 1 showed long-range correlations from H-2 (δ H 6.45) to C-1 (δ C 162.3), C-4 (δ C 121.8), and C-1a (δ C 117.5), and from Me-3 (δ H 2.07) to C-2 (δ C 117.3), C-3 (δ C 146.8), and C-4 (δ C 121.8), indicating the presence of an A ring. The HMBC correlations between H-7 (δ H 6.83) and C-5 (δ C 112.5), C-6 (δ C 164.6), C-8 (δ C 161.9), and C-8a (δ C 117.1) indicated the presence of a B ring. Meanwhile, the HMBC correlations from H-2′ (δ H 6.59) to C-1′ (δ C 162.6), C-4′ (δ C 122.2), and C-1a′ (δ C 116.0), and from Me-3′ (δ H 2.29) to C-2′ (δ C 117.7), C-3′ (δ C 148.3), and C-4′ (δ C122.2) indicated the presence of an A′ ring. The HMBC correlations from H-7′ (δ H 6.18) to C-5′ (δ C 109.7), C-6′ (δ C 166.4), C-8′ (δ C 165.7), and C-8a′ (δ C 111.5), and from OCH 3-6′ to C-6′ (δ C 166.4) indicated the presence of an B′ ring. The configuration of the C-10/C-10′ junction of the two anthronyl moieties was deduced from the HMBC correlations observed between the proton at δ H 4.14 (1H, d, J = 3.5 Hz, H-10) and C-1a (δ C 117.5), C-4 (δ C 121.8), C-5 (δ C 112.5), C-8a (δ C 117.1), and C-10′ (δ C 56.5) and between the proton at δ H 4.08 (1H, d, J = 3.5 Hz, H-10′) and C-1a′ (δ C 116.0), C-4′ (δ C 122.2), C-5′ (δ C 109.7), C-8a′ (δ C 111.5), and C-10 (δ C 57.7). The HMBC correlations between δ H 4.60 (1H, d, J = 7.5 Hz, H-1′′) and C-8 (δ C 161.9) suggested that the sugar moiety in 1 was attached at C-8. The β-pyranoside configuration was inferred from the coupling constant (3 J H-1′′, H-2′′ = 7.5 Hz) (Tian et al. 2013), while the d-glucosyl stereochemistry was determined on the basis of the enzymatic hydrolysis with β-glycoside hydrolase, followed by GC analysis of its corresponding trimethylsilylated l-cysteine adduct. Thus, the planar structure of 1 was assigned as physcion-emodin-8-O-β-d-glucopyranoside dianthrone.
The relative structure of dianthrone derivatives was unable to be confirmed by NOESY experiments (Lenta et al. 2008; Haasnoot et al. 1980; Spassov 1971). Indeed, the ROESY spectrum correlations of H-10 with H-4, H-5, H-4′ and H-5′, and H-10′ with H-4, H-5, H-4′ and H-5′ indicated that the relative structure of 1 cannot be confirmed by NOESY. Three of the four C-10/10′ diastereomers of prinoidin–emodin dianthrones (Mai et al. 2001) isomers gave two doublets (J = 3.0 Hz) for the δ H H-10 and H-10′ signals. Thus, the relative structure of dianthrone derivatives cannot be confirmed from the coupling constants between H-10 and H-10′.
The anti-conformer configuration is suggested to be neglected relative to two predominant gauche conformers with crossed rings A/B′ (I) and A′/B (II) for the cis H-10/10′ dianthrones or crossed A/A′ (III) and B/B′ (IV) for the trans H-10/10′ dianthrones (Angela et al. 2007; Ji et al. 2014). The crossed rings A/B′ (I), A′/B (II), A/A′ (III) and B/B′ (IV) shift 1H NMR peaks form the overlapped parts upfield because of their mutually-shielding effect. The H-4′/H-5 and H-4/H-5′ peaks in the crossed rings A/B′ (I) and A′/B (II) of cis H-10/10′ dianthrones shift upfield because of the shielding effects from A′/B and A/B′, respectively. The H-4′/H-4 and H-5′/H-5 peaks from the crossed rings A/A′ (III) and B/B′ (IV) of trans H-10/10′ dianthrones shift upfield because the shielding effect from A/A′ and B/B′, respectively. On the basis of above evidences, the H-4 (δ H 5.58) and H-5′ (δ H 5.45) peaks of 1 are more upfield than the H-5 (δ H 6.30) and H-4′ (δ H 6.34) peaks due to the shielding effect from A′/B (II) rings, suggesting the relative structure of 1 was confirmed as a cis H-10/10′ dianthrone (Fig. 1).
Compounds 2–4 (Fig. 1) had the same molecular formula C37H34O13 as 1, as determined by HRESI-MS, corresponding to 21 degrees of unsaturation. The IR, UV and NMR spectra of 2–4 (Tables 1, 2) were similar with those of 1. The structures were deduced from 1D NMR and 2D NMR spectra, as well as the comparison of their NMR data with those of 1. The signals of H-10 and H-10′ differed from 2 to 4. In the 1H NMR spectra of 1, 2 and 4, the H-10 and H-10′ signals all appeared as two doublets at δ H 4.14 (1H, d, J = 3.5 Hz, H-10) and 4.08 (1H, d, J = 3.5 Hz, H-10′) (1); δ H 4.57 (1H, d, J = 3.6 Hz, H-10) and 4.56 (1H, d, J = 3.6 Hz, H-10′) (2); and δ H 4.39 (1H, d, J = 3.0 Hz, H-10) and 4.35 (1H, d, J = 3.0 Hz, H-10′) (4). The 1H NMR spectrum of 3 revealed a 2H singlet assigned to both these protons at δ H 4.55 (2H s, H-10, 10′) (3). In the HMBC spectra of 1–4, the correlations from H-1″ to C-8 and from OCH 3-6′ to C-6′ were observed, suggesting that 1–4 were four C-10/C-10′ diastereomers of physcion-emodin-8-O-glucopyranoside dianthrone. A β-configuration was inferred for these compounds from the anomeric coupling constants (3 J H-1″, H-2″ >7.0 Hz), while the d-glucosyl absolute configuration was determined from enzymatic hydrolysis with β-glucosidase hydrolase. The planar structures of 2–4 were elucidated as emodin–emodin-8-O-β-d-glucopyranoside dianthrones.
The H-4/H-5′ signals of 1 and 2 are shifted to downfield due to the shielding effect from A′/B (II) rings, respectively. The H-4′/H-4 signals of 3 and 4 are also shifted to downfield due to the shielding effect from A/A′ (III) rings, respectively. These data suggest the relative structures of 1 and 2 were cis H-10/10′ dianthrones, while the relative structures of 3 and 4 were trans H-10/10′ dianthrones. Compounds 1–4 were named as polygonumnolide A1, A2, A3 and A4, respectively.
Compounds 5–7 (Fig. 1) were obtained as yellow powders. They had the same molecular formula C43H44O18, as determined by HRESI-MS, corresponding to 22 degrees of unsaturation. The IR, UV and NMR spectra of 5–7 (Tables 3, 4) were similar with those of 1–4, except they showed additional peaks assigned to an additional glucose, which indicated that 5–7 may be three C-10/C-10′ diastereomers of physcion-8-O-β-d-glucopyranoside-emodin-8-O-β-d-glucopyranoside dianthrones. The structures of these compounds were deduced from 1D NMR spectra, such as 1H NMR (Fig. 2), and 2D NMR spectra, such as 1H-1H COSY, HSQC and HMBC (Fig. 2), as well as the comparison of their NMR data with that of 1–4. The 1H NMR signals of H-10 and H-10′ differed from 5 to 7, appearing as two different signals at δ H 4.30 (1H, s, H-10) and 4.32 (1H, s, H-10′) (5), δ H 4.39 (1H, d, J = 3.0 Hz, H-10) and 4.32 (1H, J = 3.0 Hz, H-10′) (6), and a 2H singlet at δ H 4.25 (2H, s, H-10, H-10′) (7). The HMBC correlations from H-1′′ to C-8, H-1′′′ to C-8′ and from OCH 3-6′ to C-6′ in the spectra of 5–7 suggested these compounds were three C-10/C-10′ diastereomers of physcion-8-O-glucopyranoside-emodin-8-O-glucopyranoside dianthrones. Furthermore, the anomeric centers were all assigned as β-configurations from the coupling constants (3 J H-1″, H-2″ = 7.2 Hz and 3 J H-1′′′, H-2′′′ = 7.8 Hz) (5), (3 J H-1″, H-2″ = 7.8 Hz and 3 J H-1′′′, H-2′′′ = 7.2 Hz) (6) and (3 J H-1″, H-2″ = 7.8 Hz and 3 J H-1′′′, H-2′′′ = 7.2 Hz) (7). Enzymatic hydrolysis of these compounds with β-glucosidase hydrolase confirmed all the sugars were d-glucose. Thus, the planar structures of 5–7 were inferred as physcion-8-O-β-d-glucopyranoside-emodin-8-O-β-d-glucopyranoside dianthrones.
The H-4/H-5′signals of 5 and 6 are shifted to downfield due to the shielding effect from A′/B (II) rings, respectively. Additionally, the H-5′/H-5 signals of 7 are shifted to downfield due to the shielding effect from B/B′ (IV) rings. These data suggest the relative structures of 5 and 6 were cis H-10/10′ dianthrones, while the relative structure of 7 was a trans H-10/10′ dianthrone. Compounds 5–7 were named as polygonumnolide B1, B2 and B3, respectively.
Compounds 1–7 were bioassayed for cytotoxicity activity against KB tumor cell lines using taxol as the positive control (Zhang et al. 2001). Compounds 1–4 exhibited moderate cytotoxicities against the KB cell lines (Table 5).
Change history
19 December 2018
The author would like to include conflict of interest statement and Grant Numbers (81773874 and 81703665) in the acknowledgement section of the online published article. The conflict of interest statement and acknowledgement section should read as.
19 December 2018
The author would like to include conflict of interest statement and Grant Numbers (81773874 and 81703665) in the acknowledgement section of the online published article. The conflict of interest statement and acknowledgement section should read as.
References
Angela S, Xu ZL, Corina P, Christoph S, Christian C (2007) Geminal bismethylation prevents polyketide oxidation and dimerization in the benastatin pathway. Angew Chem Int Ed 46:7035–7038
Donald WC, John SE, Warwick DR (1976) Oxidation of emodin anthrone and stereochemistry of emodin biananthrone. Aust J Chem 29:1535–1548
Du L, Zhu TJ, Liu HB, Fang YC, Zhu WM, Gu QQ (2008) Cytotoxic polyketides from a marine-derived fungus Aspergillus glaucus. J Nat Prod 71:1837–1842
Gizachew A, Berhanu A, Gunther S, Helmut D (1993) Dianthrones from Senna longiracemosa. Phytochemistry 32:1273–1277
Haasnoot CAG, De Leeuw FAAM, Altona C (1980) The relationship between proton–proton NMR coupling constants and substituent electronegativities—I: an empirical generalization of the karplus equation. Tetrahedron 36:2783–2792
Ji NY, Liang XR, Sun RR, Miao FP (2014) A rule to distinguish diastereomeric dianthrones by 1H NMR. RSC Adv 4:7710–7715
Lemli J, Dequeker R, Cuveele J (1964) Recherches sur les drogues a paincipes anthraquinoniques. Planta Med 12:107–111
Lenta BN, Devkota KP, Ngouela S, Boyom FF, Naz Q, Choudhary MI, Tsamo E, Rosenthal PJ, Sewald N (2008) Anti-plasmodial and Cholinesterase inhibiting activities of some constituents of Psorospermum glaberrimum. Chem Pharm Bull 56:222–226
Lin LF, Ni BR, Lin HM, Zhang M, Li XC, Yin XB, Qu CH, Ni J (2015) Traditional usages, botany, phytochemistry, pharmacology and toxicology of Polygonum multiflorum Thunb.: a review. J Ethnopharmacol 159:158–183
Mai LP, Gueritte F, Dumontet V, Tri MV (2001) Cytotoxicity of rhamnosylanthraquinones and rhamnosylanthronese from rhamnus nepalensis. J Nat Prod 64:1162–1168
Monache GD, Rosa MC, Scurria R, Monacelli B, Pasqua G, Olio GD (1991) Metabolites from in vitro cultures of Cassia diaymobotrya. Phytochemistry 30:1849–1854
Spassov SL (1971) Nuclear magnetic resonance spectra, configuration and conformation of diastereomers—II: ethyl esters of 3-substituted 2,3-diphenylpropanoic acids: magnetic nonequivalence induced by two asymmetric centers. Tetrahedron 27:1323–1329
The State Pharmacopoeia Commission (2015 version A) The People’s Republic of China Pharmacopoeia. Chinese Medicine Science and Technology Press, Beijing, pp 175–176
Tian J, Ma QG, Yang JB, Wang AG, Ji TF, Wang YG, Su YL (2013) Hepatoprotective phenolic glycosides from Gymnema tingens. Planta Med 79:761–767
Vandenberg AJJ, Labadie RP (1981) The production of acetate derived hydroxyanthranquinones, dianthrones, naphthalenes and benzenes in tissue cultures from Rumex alpinus. Planta Med 41:169–173
Zhang PC, Wang S, Wu Y, Chen RY, Yu DQ (2001) Five new diprenylated flavonols from the leaves of Broussonetia kazinoki. J Nat Prod 64:1206–1209
Acknowledgments
The authors are grateful to the members of the analytical group of the State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China, for measuring the spectroscopic data. This Project was financially supported by the National Natural Science Foundation of China (Grant No. 81173506) and the 12th 5 Year National significant new drugs creation feature subjects-traditional Chinese medicine quality safety evaluation and risk control technology platform (2014ZX09304307-002).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
12272_2016_816_MOESM1_ESM.doc
Spectroscopic data for the new compounds (1–7) including 1H, 13C, and 2D NMR, IR and ESI-MS/MS data are provided in the supporting information
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yang, J., Yan, Z., Ren, J. et al. Polygonumnolides A1–B3, minor dianthrone derivatives from the roots of Polygonum multiflorum Thunb. Arch. Pharm. Res. 41, 617–624 (2018). https://doi.org/10.1007/s12272-016-0816-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0816-7