
Abstract
Three new xanthones, paucinervins H–J (1–3), as well as eleven known compounds (4–14), were isolated from the leaves of Garcinia paucinervis. The structures of the new compounds (1–3) were elucidated by 1D, 2D NMR spectra and HR ESIMS. In vitro antiproliferative activity against human promyelocytic leukemia HL-60 cells was tested, among which, compounds 2, 5, 6 and 7 exhibited strong growth inhibitory effects with GI50 values ranging from 1.30 to 9.08 μM, respectively. Preliminary SARs were also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Garcinia genus, belonging to the Guttiferae family, is mainly distributed in lowland rainforests of China, India, Indonesia, West and Central Africa, and Brazil. Previous chemical investigations have reported that the genus is a rich source of polyisoprenylated xanthones, benzophenones and biflavonoids. Some of them exhibit various biological activities such as antimalarial, antioxidant, anti-inflammatory, antibacterial and antitumor effects (Han and Xu 2009; Chantarasriwong et al. 2010; Hemshekhar et al. 2011). Garcinia paucinervis is a medicinally valuable species distributing in the southwest of China and used for clearing away heat and toxic materials, and dispersing swelling. The previous studies on chemical constituents of the dried leaves of G. paucinervis afforded xanthones, benzophenones, diterpenoids, depsidones, and phenolic acids. The apoptotic properties in HeLa-C3 cells and anti-tobacco mosaic virus (anti-TMV) activities of these compounds were also reported (Gao et al. 2010; Wu et al. 2013).
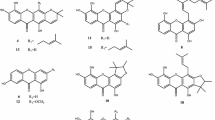
In our continuing search for antitumor natural products from the genus Garcinia (Wang et al. 2008a, b; Niu et al. 2012; Ji et al. 2012; Jing et al. 2013), the phytochemistry of G. paucinervis leaves collected from Yunnan Province of China was investigated, resulting in the isolation of fourteen compounds from the ethanol extract, including three new ones paucinervins H–J (1–3). By comparison with the published data, the known compounds were identified as 1,3,7-trihydroxy-4-prenylxanthone (4) (Lin et al. 2012), 3,4,6,8-tetrahydroxy-2-prenylxanthone (5) (Zhou et al. 2008), 1,3,5-trihydroxy-2-prenylxanthone (6) (Garcia et al. 1998), 1,3,7,8-tetrahydroxy-2-prenylxanthone (7) (Bennett et al. 1990), 1,7,8-trihydroxy-3-methoxyxanthone (8) (Xiao et al. 2001), atroviridin (9) (Kosin et al. 1998), jacareubin (10) (Zhong et al. 2009), 5,9,10-trihydroxy-2,2-dimethyl-8-(3-methyl-2-butenyl)-2H,6H-pyrano[3,2-b]xanthen-6-one (11) (Monache et al. 1984), 7,9,12-trihydroxy-2,2-dimethyl-2H,6H-pyrano[3,2-b]xanthen-6-one (12) (Nguyen et al. 2005), osajaxanthone (13) (Chang et al. 1989), and pyranojacareubin (14) (Waterman and Crichton 1980). Most of these compounds have not been isolated from G. paucinervis, except for compound 10 (Fig. 1; Gao et al. 2010). Herein, we report the structural elucidation of three new compounds, as well as the evaluation of antiproliferative activity of all obtained compounds against HL-60 cell line.

The structure of compounds 1–14 from Garcinia paucinervis
Materials and methods
General experimental procedures
The UV spectra were recorded with a Shimadzu UV-2201 spectrometer. HR ESIMS was measured on a Bruker Micro-TOFQ-Q mass spectrometer. NMR data were obtained from Bruker AV-600 NMR spectrometer using TMS as an internal standard. Silica gel (200–300 mesh) performed for column chromatography was purchased from Qingdao Ocean Chemical Factory and ODS (50 μm) was purchased from YMC Co. Ltd., Kyoto Japan. The Sephadex LH-20 was purchased from GE Healthcare. A Shimadzu SPD-20A series equipped with an YMC C18 column (250 × 20 mm, 5 μm) was used for HPLC analysis and semi-preparation. 5-fluorouracil (5-Fu, purity >99 % by HPLC) was purchased from Aladdin. All the organic solvents were purchased from Yuwang Chemicals Industries, Ltd., China.
Plant material
Garcinia paucinervis was collected from Yunnan province, China, in March 2012 and was identified by Mr. Yu Chen (Xishuangbanna Tropical Botanic Garden of the Chinese Academy of Sciences, People’s Republic of China). A voucher sample (JSL-1203) was deposited in the Department of Natural Products Chemistry, Shenyang Pharmaceutical University, Shenyang, China.
Extraction and isolation
The air-dried leaves of G. paucinervis (6.5 kg) were extracted with ethanol (3 × 8 L 95:5, v/v) for three times (3 h, each) at room temperature. The extract was concentrated under reduced pressure to give black-brown gum of 1412 g. The residue was suspended in water and then partitioned with petroleum ether (3 × 6 L), CH2Cl2 (3 × 6 L) and n-BuOH (3 × 6 L), successively. The CH2Cl2 soluble part (350 g) was subjected to silica gel column chromatography (CC, i.d. 10 × 120 cm) eluting with a PE/acetone gradient system from 100:0 to 0:100 to give six fractions Fr.A ~ F based on their TLC characteristics. Fr.C (22 g) was decolorized by D101 macroporous resin. The 90 % methanol part was performed on silica gel CC (i.d. 4 × 50 cm) eluting with a PE/EtOAc gradient system from 100:0 to 0:100 to give five subfractions (Fr.C1 ~ Fr.C5). Fr.C1 (3 g) was submitted to Sephadex LH-20 CC (i.d. 1 × 55 cm) eluting with methanol and followed by semi-preparative HPLC (YMC C18 column, 250 × 20 mm, 5 μm, over 60 min at a flow rate of 2 mL/min) using MeOH/H2O 82:18 as the mobile phase to afford compounds 2 (2.5 mg, tR = 21 min), 3 (2.3 mg, tR = 24 min) and 7 (5 mg, tR = 26 min). Fr.C3 (2 g) was separated under the same conditions to obtain 10 (3.1 mg). Fr.D (30 g) was decolorized by the same method as Fr.C and then subjected to ODS CC (i.d. 5.5 × 60 cm) eluting with a MeOH/H2O gradient system from 60:40 to 90:10 to give five major subfractions (Fr.D1 ~ Fr.D5). Fr.D1 (1 g) was applied to Sephadex LH-20 CC (i.d. 1 × 55 cm) and then purified by semi-preparative HPLC (YMC C18 column, 250 × 20 mm, 5 μm, over 60 min at a flow rate of 2 mL/min) using MeOH/H2O 78:22 as the mobile phase to yield 4 (3.6 mg, tR = 27 min), 5 (4.2 mg, tR = 31 min), 8 (4.8 mg, tR = 24 min) and 9 (4.0 mg, tR = 33 min). Fr.D3 (2 g) was also purified under the same conditions using MeOH/H2O 73:27 as the mobile phase yielding 1 (2.2 mg, tR = 14 min) and 13 (6.5 mg, tR = 17 min). Fr.E (21 g) and Fr.F (33 g) were isolated respectively under same conditions as Fr.C and finally yielded 6 (3.4 mg) from Fr.E and 4 (3.7 mg), 11 (2.8 mg), 12 (3.2 mg) and 14 (3.6 mg) from Fr.F.
Paucinervin H (1): yellow amorphous powder; UV (MeOH) λ max: 258, 323, 329 nm; 1H and 13C NMR (600 and 150 MHz, measured in DMSO-d 6) data see Table 1; HR ESIMS: m/z = 449.1570 [M + Na]+ (calcd for C24H26O7Na, 449.1576).
Paucinervin I (2): yellow amorphous powder; UV (MeOH) λ max: 276 nm; 1H and 13C NMR (600 and 150 MHz, measured in DMSO-d 6) data see Table 2; HR ESIMS: m/z = 395.1486 [M + H]+ (calcd for C23H23O6, 395.1495).
Paucinervin J (3): yellow amorphous powder; UV (MeOH) λ max: 258, 323, 329 nm; 1H and 13C NMR (600 and 150 MHz, measured in acetone-d 6) data see Table 1; HR ESIMS: m/z = 365.0988 [M + Na]+ (calcd for C19H18O7Na, 365.1001).
Cell culture and in vitro antiproliferative activity assay
HL-60 cells were plated in a 96-well plate. After 24 h spreading, various concentrations of obtained compounds were added into each well at 37 °C incubating for 4 days. The cultures were then treated with MTT solution for an additional 4 h. After discarding culture medium, the cells were dissolved in 200 μL DMSO and the optical density (OD) at 570 nm was measured by a microplate reader spectrophotometer. The concentration of a compound (purity >98 % by HPLC) inhibiting half of the cell growth was calculated, using 5-Fu (80 mM/L) as a positive control. The results were obtained from three independent experiments carried out in duplicate.
Results and discussion
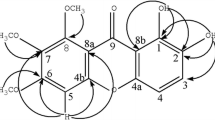
Compound 1 was obtained as yellow amorphous powder. Its molecular formula was established as C24H26O7 by HR ESIMS at m/z 449.1570 [M + Na]+ (cacld. 449.1576). The UV absorptions at 258, 323 and 329 nm indicated that 1 was a xanthone derivative (Ito et al. 2003). 1H and 13C NMR data for 1 (Table 1) were almost identical to those for nigrolineaxanthone E, which was previously isolated from G. paucinervis (Rukachaisirikul et al. 2003). The main differences in the 1H NMR spectrum were a non-oxygenated methylene at δ 2.75 (2H, m, H-1′), an oxygenated methine at δ 4.36 (1H, t, J = 7.1 Hz, H-2′), a terminal double bond at δ 4.67 and 4.61 (each 1H, brs, H-4′), and a methyl group at δ 1.72 (3H, s, H-5′), which suggested the occurrence of a different side chain of 2-hydroxy-3-methylbut-3-enyl group at C-2. The 13C NMR chemical shifts of above group were also observed at δ 148.5 (C-3′), 109.7 (C-4′), 72.7 (C-2′), 30.0 (C-1′) and 17.3 (C-5′). Furthermore, the HMBC correlations (Table 1; Fig. 2) confirmed that compound 1 was 1,5,6-trihydroxy-2-(2-hydroxy-3-methylbut-3-enyl)-4-(1,1-dimethylprop-2-enyl)-3-methoxyxanthone and named paucinervin H. 1 showed no significant absorption in CD spectrum (Fig. S5) and lacked of optical activity, which was indicative of its racemic nature (Fig. 1).
Compound 2 was isolated as yellow amorphous powder. Its molecular formula C23H22O6 was deduced by its HR ESIMS (m/z 395.1486 [M + H]+). The UV spectrum showed absorption maxima at 276 nm. The 1H and 13C NMR data (Table 2) assigned by the HMBC spectra, suggested that 2 were a prenylated pyranoxanthone. In the HMBC spectrum (Table 2; Fig. 2), a chelated hydroxyl proton at δ 13.03 showed the long-range correlations with C-1 (δ 160.4), C-2 (δ 97.7) and C-9a (δ 101.2), assigning the aromatic proton (δ 6.20) to H-2. The significantly upfield chemical shifts of H-2 and C-2 revealed that both of ortho-positions were oxygen-substituted. The long-range correlation of one olefinic proton at δ 6.56 of a cis-double bond with a carbon at δ 146.2 (C-3) was suggestive of the location of dimethylpyran ring at C-3 and C-4 of the xanthone nucleus. Another singlet aromatic proton at δ 7.34 was assigned to be H-8 by its significant long-range correlation with the carbonyl carbon at δ 179.4 (C-9). The prenyl group was linked to C-7 due to the long-range correlations observed from the H-1′′ methylene protons at δ 3.42 to C-6 (δ 154.1), C-7 (δ 122.6) and C-8 (δ 113.7). The remaining hydroxyl groups were then attached to C-5 and C-6 respectively based on its molecular formula. Thus, the structure of compound 2 was determined as 6,10,11-trihydroxy-3,3-dimethyl-9-(3-methylbut-2-enyl)-pyran[2,3-c]xanthen-7(3H)-one and named paucinervin I.

Key HMBC correlations of compounds 1–3
Compound 3 was obtained as yellow amorphous powder with the molecular formula C19H18O6 determined by a [M + Na]+ quasi-molecular ion peak at m/z 365.0988 in its HR ESIMS. It exhibited UV absorption bands similar to those of compound 1 at 258, 323 and 329 nm, which was suggestive of a xanthone derivative (Ito et al. 2003). Its 1H and 13C NMR data were showed in Table 1. The aromatic proton at δ 7.58 was attributed to H-8 on the basis of its downfield chemical shift as well as the HMBC (Table 1; Fig. 2) correlation with the carbonyl carbon at δ 180.9. Based on coupling constants, the aromatic protons at δ 7.34 and 7.50 were assigned to H-6 and H-5, respectively. The long-range correlations of H-5 with C-8a (δ 121.9) and C-7 (δ 150.5) showed that C-7 was oxygenated carbon. The prenyl group was attached at C-2 deduced from the long-range correlations of the chelated hydroxyl group at δ 13.10 with δ 156.9 (C-1), 111.1 (C-2) and 102.4 (C-9a) and the methylene protons at δ 3.37 (H-1′) with δ 156.9 (C-1), 111.1 (C-2), 123.5 (C-2′) and 131.4 (C-3′). The significant downfield shift of the methoxy carbon at δ 61.8 indicated that both of the ortho-positions of this methoxy group are oxygen-substituted (Rukachaisirikul et al. 2003). Thus, this methoxyl group was assigned to be located at C-4 which was supported by the long-range correlation between the methoxy protons at δ 3.91 and an oxyquaternary carbon at δ 128.2 (C-4) located at the center of three adjacent oxygenated carbons. Similarly, 3,7-dihydroxy substitution was determined by the molecular formula C19H18O6 of 3. 3 showed the similar NMR data of A ring to garcinenone X (Ji et al. 2012) and of B ring to 1,3,7-trihydroxy-4-prenylxanthone (Lin et al. 2012), supporting the above structure elucidation. Thus, compound 3 was established as 1,3,7-trihydroxy-4-methoxy-2-prenylxanthone and was named paucinervin J.
All the isolated compounds were tested for the growth inhibitory effects against HL-60 cell line using the MTT assay. All the compounds showed inhibitory activities with GI50 values ranging from 1.30 to 49.52 μM, which are listed in Table 3. Among them, compounds 2, 5, 6 and 7, each possessing a prenyl group, exhibited more potent in vitro antiproliferative effects with GI50 values of 1.30, 4.97, 6.06 and 9.08 μM, respectively. The result is consistent with our previous report on the xanthones from G. bracteata (Niu et al. 2012), that the prenyl group is favorable for antiproliferative activity against tumor cells. Compounds 8, 9, 10, 12, 13 and 14, without prenyl group, exhibited weaker cytotoxic activity, among which, compound 8 with a GI50 value of 49.52 μM is the weakest one. It is noteworthy that compound 2 with angular pyranoxanthone skeleton demonstrated more potent antiproliferative effect than the corresponding linear compound (11), indicating that the activity was influenced by the pyran-fused position. And the antiproliferative activity of 2 with a GI50 value of 1.30 μM was stronger than that of positive control 5-FU (GI50 value of 2.37 μM).
The chemical investigation of G. paucinervis leaves led to the isolation of three new (paucinervins H-J, 1–3) and eleven known compounds (4–14). Most of them were firstly obtained from G. paucinervis. The structures of 1–3 were elucidated on the basis of their spectroscopic data (HR ESIMS, 1H NMR, 13C NMR, HMBC and UV) and by comparison with the literature data. The results of in vitro antiproliferative activity assay showed that 2, 5, 6 and 7, each with the prenyl group, exhibited stronger growth inhibitory effects against HL-60 cells, with GI50 values of 1.30, 4.97, 6.06 and 9.08 μM, respectively. New compound 2 was the most potent one showed a GI50 value inferior to that of 5-FU against HL-60 cells, which deserved further investigation. While, compounds 8, 9, 10, 12, 13 and 14, without prenyl group, exhibited weaker cytotoxic activity. Preliminary SAR of these xanthones was also summarized.
Supporting information
HR ESIMS, 1H NMR, 13C NMR, HMBC and UV spectra of compounds 1–3 are available as Supporting Information.
References
Bennett GJ, Lee HH, Lee LP (1990) Synthesis of minor xanthones from Garcinia mangostana. J Nat Prod 53:1463–1470
Chang CH, Lin CC, Hattori M, Namba T (1989) Four prenylated xanthones from Cudrania cochinchinensis. Phytochemistry 28:595–598
Chantarasriwong O, Batova A, Chavasiri W, Theodorakis AE (2010) Chemistry and biology of the caged Garcinia xanthones. Chem-A Eur J 16:9944–9962
Gao XM, Yu T, Lai FS, Zhou Y, Liu X, Qiao CF, Song JZ, Chen SL, Luo KQ, Xu HX (2010) Identification and evaluation of apoptotic compounds from Garcinia paucinervis. Bioorg Med Chem 18:4957–4964
Garcia CDA, Young MCM, Marston A, Wolfender JL, Hostettmann K (1998) Xanthones, triterpenes and a biphenyl from Kielmeyera coriacea. Phytochemistry 47:1367–1374
Han QB, Xu HX (2009) Caged Garcinia xanthones: development since 1937. Curr Med Chem 16:3775–3796
Hemshekhar M, Sunitha K, Santhosh MS, Devaraja S, Kemparaju K, Vishwanath BS, Niranjana SR, Girish KS (2011) An overview on genus garcinia: phytochemical and therapeutical aspects. Phytochem Rev 10:325–351
Ito C, Itoigawa M, Takakura T, Ruangrungsi N, Enjo F, Tokuda H, Nishino H, Furukawa H (2003) Chemical constituents of Garcinia fusca: structure elucidation of eight new xanthones and their cancer chemopreventive activity. J Nat Prod 66:200–205
Ji F, Li ZL, Liu GY, Niu SL, Zhao N, Liu XQ, Hua HM (2012) Xanthones with antiproliferative effects on prostate cancer cells from the stem bark of Garcinia xanthochymus. Nat Prod Commun 7:53–56
Jing WY, Jiang C, Ji F, Hua HM, Li ZL (2013) Chemical constituents from the stem barks of Garcinia multiflora. J Asian Nat Prod Res 15:1152–1157
Kosin J, Ruangrungsi N, Ito C, Furukawa H (1998) A xanthone from Garcinia atroviridis. Phytochemistry 47:1167–1168
Lin CF, Chen YJ, Huang YL, Chiou WF, Chiu JH, Chen CC (2012) A new auronol from Cudrania cochinchinensis. J Asian Nat Prod Res 14:704–707
Monache GD, Monache FD, Waterman PG, Crichton EG, Lima RAD (1984) Minor xanthones from Rheedia gardneriana. Phytochemistry 23:1757–1759
Nguyen LH, Venkatraman G, Sim KY, Harrison LJ (2005) Xanthones and benzophenones from Garcinia griffithii and Garcinia mangostana. Phytochemistry 66:1718–1723
Niu SL, Li ZL, Ji F, Liu GY, Zhao N, Liu XQ, Jing YK, Hua HM (2012) Xanthones from the stem bark of Garcinia bracteata with growth inhibitory effects against HL-60 cells. Phytochemistry 77:280–286
Rukachaisirikul V, Ritthiwigrom T, Pinsa A, Sawangchote P, Taylor WC (2003) Xanthones from the stem bark of Garcinia nigrolineata. Phytochemistry 64:1149–1156
Wang LL, Li ZL, Song DD, Sun L, Pei YH, Jing YK, Hua HM (2008a) Two novel triterpenoids with antiproliferative and apoptotic activities in human leukemia cells isolated from the resin of Garcinia hanburyi. Planta Med 74:1735–1740
Wang LL, Li ZL, Xu YP, Liu XQ, Pei YH, Jing YK, Hua HM (2008b) A new cytotoxic caged polyprenylated xanthone from the resin of Garcinia hanburyi. Chin Chem Lett 19:1221–1223
Waterman PG, Crichton EG (1980) Xanthones and biflavonoids from Garcinia densivenia stem bark. Phytochemistry 19:2723–2726
Wu YP, Zhao W, Xia ZY, Kong GH, Lu XP, Hu QF, Gao XM (2013) Three novel xanthones from Garcinia paucinervis and their anti-TMV activity. Molecules 18:9663–9669
Xiao H, Lu Y, Chen ZN, Liu GM, Qian JF (2001) New xanthones from Swertia decora. J Chin Pharm Sci 10:172–174
Zhong FF, Chen Y, Wang P, Feng HJ, Yang GZ (2009) Xanthones from the bark of Garcinia xanthochymus and their 1,1-diphenyl-2-picrylhydrazyl radical-scavenging activity. Chin J Chem 27:74–80
Zhou Y, Han QB, Song JZ, Qiao CF, Xu HX (2008) Characterization of polyprenylated xanthones in Garcinia xipshuanbannaensis using liquid chromatography coupled with electrospray ionization quadrupole time-of-flight tandem mass spectrometry. J Chromatogr A 1206:131–139
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (31570350, 21502121), Project Funded by China Post Doctoral Science Foundation (2015M570258), General Scientific Research Projects of Department of Education in Liaoning Province (L2014382), Young Teachers’ Scientific Research Fund Project of Shenyang Pharmaceutical University (QNJJ 2013501) and Career Development Support Plan for Young and Middle-aged Teachers in Shenyang Pharmaceutical University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Da-Hong Li and Chen-Xi Li have contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, DH., Li, CX., Jia, CC. et al. Xanthones from Garcinia paucinervis with in vitro anti-proliferative activity against HL-60 cells. Arch. Pharm. Res. 39, 172–177 (2016). https://doi.org/10.1007/s12272-015-0692-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-015-0692-6