
Abstract
Cardiomyopathy has variable penetrance. We analyzed age and sex-related genetic differences in 1,397 cardiomyopathy patients (Ontario, UK) with whole genome sequencing. Pediatric cases (n = 471) harbored more deleterious protein-coding variants in Tier 1 cardiomyopathy genes compared to adults (n = 926) (34.6% vs 25.9% respectively, p = 0.0015), with variant enrichment in constrained coding regions. Pediatric patients had a higher burden of sarcomere and lower burden of channelopathy gene variants compared to adults. Specifically, pediatric patients had more MYH7 and MYL3 variants in hypertrophic cardiomyopathy, and fewer TTN truncating variants in dilated cardiomyopathy. MYH7 variants clustered in the myosin head and neck domains in children. OBSCN was a top mutated gene in adults, enriched for protein-truncating variants. In dilated cardiomyopathy, female patients had a higher burden of z-disc gene variants compared to males. Genetic differences may explain age and sex-related variability in cardiomyopathy penetrance. Genotype-guided predictions of age of onset can inform pre-test genetic counseling.
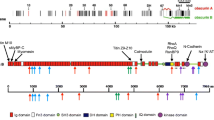
Graphical Abstract
Pediatric cardiomyopathy patients were more likely to be genotype-positive than adults with a higher burden of variants in MYH7, MYL3, TNNT2, VCL. Adults had a higher burden of OBSCN and TTN variants. Females with dilated cardiomyopathy (DCM) had a higher burden of z-disc gene variants compared to males.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiomyopathies are among the most common genetic cardiac disorders and the leading cause of heart failure and sudden cardiac death [1]. Phenotypically, there are five subtypes—dilated (DCM), hypertrophic (HCM), restrictive (RCM), left ventricular non-compaction (LVNC) and arrhythmogenic (ACM) cardiomyopathy. They can be sporadic or familial, and usually have an autosomal dominant inheritance [2,3,4,5]. Variants in over 100 genes have been associated with cardiomyopathy, with considerable overlap in genes involved in causing different cardiomyopathy subtypes [4, 6, 7].
While some individuals manifest clinical cardiomyopathy in childhood, others do not manifest disease until adulthood [8,9,10,11]. Studies have also reported sex-related differences in the incidence and severity of cardiomyopathy [12,13,14,15]. While age-related penetrance, lifestyle factors, and hormonal differences can explain part of the clinical variability by age and sex [16,17,18], a systematic comparison of childhood versus adult-onset cardiomyopathy, or of cardiomyopathy in females versus males, using comprehensive genomic evaluation is important. Marston et al. reported a higher yield of sarcomere gene variants in pediatric versus adult HCM [9]. However, their study was limited to patients with HCM, was confounded by the variability in the sizes of gene panels used for clinical testing across patients, only focused on sarcomere gene variants, and did not analyze sex-related differences. Whole genome sequencing can overcome some of these limitations by permitting a standardized and comprehensive exploration of all relevant genes across different cohorts and also allows an exploration of not just single nucleotide variants but also of structural variants.
The goal of our study was to use whole genome sequencing to evaluate age and sex-related differences in the genetics of cardiomyopathy by comparing the frequency and distribution of rare, deleterious variants in known cardiomyopathy genes and within constrained regions of these genes between patients with childhood and adult-onset cardiomyopathy, and between affected males and females.
Results
The study cohort comprised 1,397 unrelated primary cardiomyopathy patients with whole genome sequencing recruited through (i) the multi-institutional Ontario province-wide Heart Centre Biobank Registry (n = 236), and (ii) the 100,000 Genomes Project accessed through the Genomics England Clinical Interpretation Partnership (version 8) (n = 1,161) [19,20,21,22,23]. Patients with secondary causes of cardiomyopathy (syndromic, neuromuscular, metabolic or mitochondrial, congenital heart disease) were excluded. 471 patients were pediatric (< 18 years old at diagnosis) (236 from the Ontario biobank, 235 from Genomics England), and 926 were adults (all from Genomics England); 871 (62%) were male. By subtype, 768 (55%) had HCM, 435 (31%) had DCM, 116 (8%) had ACM, 58 (4%) had LVNC, and 20 (1%) had RCM. The cohort characteristics are described in Table 1. The pediatric cohort had a higher proportion of DCM, LVNC and RCM compared to the adult cohort. Written informed consent was obtained from all biobank participants and/or their parents or legal guardians and the research protocol was approved by the Institutional Research Ethics Boards at all participating sites. The full methods are available as Supplementary Material.
Variant burden in pediatric versus adult cardiomyopathy
We mapped protein-coding single nucleotide variants, indels and copy number variants to 78 cardiomyopathy candidate genes (30 Tier 1, 48 Tier 2) with autosomal dominant inheritance that were manually curated based on published evidence and representation on commercial gene panels (Supplementary Table S1) [24,25,26,27,28].
Higher frequency of deleterious variants in pediatric cardiomyopathy
Overall, 588 patients (42.1% of the cohort) harbored one or more rare deleterious variants in 72 genes. A majority i.e., 403 (28.8% of the overall cohort) harbored variants in 30 Tier 1 genes (34.6% in pediatric, 25.9% in adults, p = 0.0015) (Online Abstract Figure). When analyzed by cardiomyopathy subtypes, a higher proportion of pediatric HCM cases harbored a deleterious variant compared to adult HCM (34.% vs 23.1% respectively, p = 1.5 × 10–3); this difference was not seen in DCM cases. There was no significant difference in the proportion of patients harboring multiple deleterious variants between pediatric (3.8%) and adult (2.7%) cases (p = 0.33). The list of variants is provided in Supplementary Table S2.
Higher variant constrained coding region (CCR) scores in pediatric cardiomyopathy
We obtained the CCR score for all deleterious variants identified in cardiomyopathy genes in our study cohort and in gnomAD. Variant CCR scores were compared between cases and reference control genomes available through the Genome Aggregation Database (gnomAD v2.1) (n = 125,748) using Kolmogorov–Smirnov (KS) two-tailed test. As expected, deleterious variants identified in cardiomyopathy patients were in more constrained coding regions compared to those in the gnomAD reference population i.e., had higher CCR scores (KS test p < 2.2 × 10–16) (Fig. 1a, c). This difference was also seen in the subset of Tier 1 genes (KS test p < 2.2 × 10–16) (Fig. 1b, d). For genes harboring deleterious variants, we generated variant CCR score density maps (Figs. 2a-d) and compared CCR score probability distribution using KS two-tailed test (Figs. 2e-h). Variant CCR scores across all cardiomyopathy genes were higher in pediatric compared to adult cases (KS test p = 0.0025) and also when limited to variants in Tier 1 genes (KS test p = 0.0047). CCR scores of variants in sarcomere genes were also higher in pediatric versus adult cases (KS test p = 0.0048). CCR scores of variants in non-sarcomere genes did not differ between pediatric and adult cases (KS test p = 0.096). Overall, these findings suggest that those with early onset disease were more likely to harbor variants within highly constrained coding regions of sarcomere genes.

CCR score of deleterious variants in cases and controls. (a, b) Density plots of CCR scores (blue = cardiomyopathy; pink = gnomAD), and (c, d) Empirical cumulative probability distribution of CCR scores of deleterious variants identified in cardiomyopathy cases (n = 1397) (green) and in gnomAD reference controls (n = 125,748) (orange). Variant CCR scores were higher in cardiomyopathy cases compared to gnomAD reference controls across all cardiomyopathy genes (KS test p < 2.2 × 10–16) and in the subset of Tier 1 genes (KS test p < 2.2 × 10–16). CCR, constrained coding regions; CMP, cardiomyopathy; gnomAD, Genome Aggregation Database; KS, Kolmogorov–Smirnov test

CCR score of deleterious variants stratified by age. (a-d) Density plots of CCR scores (blue = pediatric; pink = adult cardiomyopathy), and (e–h) Empirical cumulative probability distribution of CCR scores of deleterious variants identified in pediatric (n = 471) (orange) and adult cardiomyopathy cases (n = 921) (green). Variant CCR scores were higher in pediatric versus adult cardiomyopathy cases across all cardiomyopathy genes (KS test p = 0.0025), in the subset of Tier 1 genes (p = 0.0047) and in sarcomere genes (p = 0.0048), but not different in non-sarcomere genes (p = 0.096). CCR, constrained coding regions; CMP, cardiomyopathy; KS, Kolmogorov–Smirnov test
Affected genes in pediatric and adult cardiomyopathy
Single nucleotide variants and indels
For each gene, we compared the proportion of patients harboring at least one deleterious variant in the gene. Figure 3a-c shows the proportion of pediatric and adult patients harboring deleterious variants by gene and by variant types i.e., missense and loss-of-function (LoF). Since there was a difference in cardiomyopathy subtypes between pediatric and adult cases, we analyzed genetic differences by cardiomyopathy subtype. Figures 3d-f show volcano plots by cardiomyopathy subtype for differentially mutated genes between pediatric and adult cases. Pediatric cases had a higher variant burden in MYH7 (OR 3.58, p = 1.2 × 10–6) and MYL3 (OR 17.8, p = 0.013) but a lower variant burden in OBSCN (OR 0.38, p = 0.002). These differences were primarily seen in HCM cases. TTN truncating variants were more frequent in adult DCM cases compared to childhood DCM cases (Fig. 3d, Supplementary Table S3). Although OBSCN is not currently considered a Tier 1 gene for cardiomyopathy, we found a significantly higher burden of high confidence OBSCN LoF variants in our overall cohort compared to gnomAD reference controls (OR = 1.72, CI 1.01–2.75, p = 0.028) (Supplementary Table S4). This enrichment was seen primarily in adult cardiomyopathy patients compared to the gnomAD reference population (p = 0.033). This enrichment of LoF OBSCN variants was not seen in pediatric cardiomyopathy patients (p = 0.41 vs gnomAD) (Supplementary Table S4). Another gene that emerged with a significant variant burden in the overall cohort was VCL. While overall accounting for fewer than 1% of cases, LoF variants in VCL were only seen in children, not in adults (OR = 13.8, p = 0.038). Finally, when genes were collapsed into functional gene categories, pediatric HCM patients had a higher burden of variants in sarcomere genes compared to adult HCM, while pediatric DCM patients had a lower burden of variants in channelopathy genes compared to adults (Fig. 4, Supplementary Table S5).

Genes harboring deleterious variants stratified by age. Bar plots showing genes harboring deleterious variants in patients with (a) any cardiomyopathy (n = 1397), (b) only hypertrophic cardiomyopathy (n = 768), and (c) only dilated cardiomyopathy (n = 435). *p < 0.05 between pediatric and adult patients by Fisher test. (d-f) Volcano plots showing log odds ratio of the frequency of variants between pediatric and adult patients with (d) any cardiomyopathy, (e) hypertrophic cardiomyopathy only, and (f) dilated cardiomyopathy only. MYH7, TNNT2, MYL3 and VCL were more frequently mutated in pediatric patients, and TTN and OBSCN were more frequently mutated in adult patients. (orange bar = missense variants, green bar = loss of function variants). NS, no significant difference (grey); log2 OR, genes meeting log2 (odds ratio) > 1 between pediatric and adult (green) (vertical lines); p, genes meeting p < 0.05 between pediatric and adult (blue) (horizontal line); p & log2 OR—genes meeting p < 0.05 with at least 1.5 odds ratio (red)

Gene categories harboring rare deleterious variants stratified by age. The forest plot shows the log odds ratio comparing proportion of variant carriers between 471 pediatric and 926 adult patients for different gene categories across cardiomyopathy subtypes. Sarcomere genes were more frequently mutated in pediatric HCM cases while channelopathy genes were more frequently mutated in adult DCM patients. Filled circles indicate significant differences between pediatric and adult patients (p < 0.05). CMP, cardiomyopathy (black); DCM, dilated cardiomyopathy (blue), HCM, hypertrophic cardiomyopathy (red)
We performed de novo variant calling in cardiomyopathy genes in a subset of 125 probands (95 pediatric and 30 adult) with complete trio WGS data. Deleterious de novo variants in known genes (MYH7, TNNI3, TPM1, TNNT2, TTN, DES, RYR2 and NF1) were identified in 11 (12%) pediatric probands but no de novo variants were identified in adult-onset cardiomyopathy (OR 8.3; p = 0.042) although the analysis was limited by the relatively small number of complete trio data.
Structural variants
Whole genome sequencing provides an opportunity to explore additional deleterious variant types that standard panel and exome sequencing have a limited capacity to detect. We investigated whether structural variants, including copy number losses or gains, inversions, or insertions, affected genes that were highly mutated between pediatric and adult cardiomyopathy patients i.e., MYH7, MYL3, OBSCN, TTNT2, TTN, and VCL. This identified six additional predicted deleterious copy number deletions in OBSCN and TTN, none of which have been previously reported in the reference population. Of these, three were also deemed pathogenic by the American College of Medical Genetics and Genomics criteria (Supplementary Table S6).
Variant location across protein domains by age
We used goodness of fit test to determine if deleterious SNVs and indels were uniformly distributed within protein domains of genes or clustered in specific protein domains within genes. This was analyzed only for the top mutated genes in the overall cohort. Figure 5 shows the spatial distribution of deleterious variants in MYH7, MYBPC3, TTN, and OBSCN against the transcript count index [29]. Deleterious variants were non-uniformly distributed in MYH7 in pediatric patients with variants clustering within the myosin head and neck domains (KS test p = 8.4 × 10–4). In adults, variants were also predominantly in the myosin head and neck domains but were also seen in the tail domain similar to the reference population (KS test, p = 0.058) (Fig. 5a). For MYBPC3, variants clustered within the C5, C7, and C10 domains with no difference between children and adults (Fig. 5b). TTN truncating variants were non-evenly distributed with clustering in the A-band (p = 3.4 × 10–4) in adult DCM patients (p = 3.5 × 10–3) [29,30,31], but not in pediatric cases (p = 0.49) (Fig. 5c). OBSCN deleterious variants were non-evenly distributed and clustered around the protein kinase domain of the gene in pediatric patients only (p = 0.0088) (Fig. 5d). When analyzed by cardiomyopathy subtypes, MYH7 variants in HCM patients clustered within myosin head and neck regions in both pediatric and adult HCM patients but were rare in the head domain in DCM patients (Supplementary Figure S1). Similarly, TTN truncating variants clustered within the A-band in both pediatric and adult DCM patients, with non-random distribution in adult DCM (Supplementary Figure S2c).

Location of deleterious variants within protein domains. Each protein is linearly depicted with uniprot domain information. Non-random distribution of variants within each protein was assessed using Kolmogorov–Smirnov goodness-of-fit test. Light salmon bars show the constrained coding region scores across the entire protein. (a) MYH7 showing head, neck and rod/tail regions. Unlike variants in adult patients, deleterious variants in pediatric patients were non-uniformly distributed with clustering within head and neck domains and fewer variants in the tail domain (p = 0.00084 vs gnomAD distribution). (b) MYBPC3 showing the immunoglobulin and fibronectin type 3 domains. Deleterious variants clustered in C5, C7, and C10 domains (p = 0.023), and this distribution did not differ between pediatric (n = 471) and adult (n = 926) patients. (c) TTN showing the four regions and all domains. The light blue bars under titin show the transcript count index. TTN truncating variants clustered in the A-band domain with non-uniform distribution in adult patients (p = 0.0035). (d) OBSCN domains. Deleterious variants clustered around the protein kinase domains of OBSCN in pediatric patients (p = 0.0088). CCR, Constrained coding region; TCI, transcript count index
Variant burden by sex
Of 526 females, 228 (43%) were genotype positive, and of 871 males, 355 (41%) were genotype-positive (p = 0.342). There were no sex-related differences in variant CCR scores in gnomAD reference genomes (p = 0.51) (Fig. 6a, d). However, in the cardiomyopathy cohort, the deleterious variant CCR scores were higher in females compared to males. This was true of variants in all cardiomyopathy genes (p = 0.0025) (Fig. 6b, e) as well as for variants in Tier 1 genes (p = 0.019) (Fig. 6c, f). There was no difference in variant burden between sexes across all cardiomyopathies, or in HCM and DCM subtypes (Fig. 7a; Supplementary Table S7). At the gene category level however, variants in z-disc genes were enriched in female DCM patients compared to male DCM patients (p = 0.027) (Fig. 7b; Supplementary Table S8).

CCR score of deleterious variants in male and female cases and controls. (a-c) Density plots of deleterious variant CCR scores (blue = male, pink = female), and (d-f) Empirical cumulative probability distribution of deleterious variant CCR scores in 1397 cardiomyopathy cases (871 males, 526 females) and in 125,748 gnomAD reference population stratified by sex (green = male; orange = female). There was no difference in variant CCR scores between males and females (a, d) for all genes in gnomAD reference population (p = 0.92), (b, e) for all genes in cardiomyopathy cases (KS test p = 0.068), and (c, f) for Tier 1 genes in cardiomyopathy cases (p = 0.16). CCR, constrained coding regions; CMP, cardiomyopathy; gnomAD, Genome Aggregation Database

Genes and gene categories harboring deleterious variants stratified by sex. (a) Bar plots showing no difference in burden of rare deleterious variants in cardiomyopathy genes between male and female patients (871 males, 526 females) across all cardiomyopathy types, (b) in HCM patients, and (c) in DCM patients (orange = missense variants, green = loss of function variants). (d) The forest plot shows the log odds ratio comparing proportion of variant carriers between male and female patients across different gene categories in DCM, HCM and overall cohort. Z-disc genes were enriched for variants in female compared to male DCM patients. Filled circles indicate significant differences between male and female patients (p < 0.05). CMP, cardiomyopathy (black); DCM, dilated cardiomyopathy (blue), HCM, hypertrophic cardiomyopathy (red); LoF, loss of function
Variant location across protein domains by sex
Figure 8 shows the spatial distribution of deleterious variants in MYH7, MYBPC3, TTN, and OBSCN. MYH7 deleterious variants clustered within myosin head and neck domain in male and female patients but this was significant only in males (KS test p = 0.0017 in males, p = 0.051 in females) likely due to lower numbers of female patients (Fig. 8a). MYBPC3 deleterious variants were evenly distributed within the protein domains in both male and female patients (Fig. 8b). TTN truncating variants were located in the A-band of TTN with uneven distribution in male patients (KS test p = 0.01) (Fig. 8c). By cardiomyopathy subtype, MYH7 deleterious variants clustered within myosin head and neck regions (Supplementary Fig. 3a) and TTN truncating variants clustered within the A-band zone in male and female patients (Supplementary Fig. 4).

Location of deleterious variants within protein domains stratified by sex. Each protein is linearly depicted with uniprot domain information. Non-random distribution of variants within each protein was assessed using Kolmogorov–Smirnov goodness-of-fit test. Light salmon bars show the constrained coding region scores across the entire protein. (a) MYH7 showing head, neck and rod/tail regions. Deleterious variants were non-uniformly distributed with clustering within head and neck domains and fewer variants in the tail domain. (b) MYBPC3 showing the immunoglobulin and fibronectin type 3 domains. Deleterious variant distribution did not differ between male and female patients. (c) TTN showing the four regions and all domains. The light blue bars under titin show the transcript count index. TTN truncating variants clustered in the A-band with non-uniform distribution in male patients (p = 0.011). (d) OBSCN LoF variants were uniformly distributed across protein domains in male and female patients. CCR, Constrained coding region; TCI, transcript count index; LoF, loss of function
Discussion
Our study provides new insights into the genetic architecture of cardiomyopathy. We showed major genetic differences by age of onset of disease and modest differences by sex. The use of whole genome sequencing in this context provided a standardized comparison between groups without being confounded by variable sizes of gene panels used in clinical testing. This helped us identify differences in variant burden by cardiomyopathy type in not only sarcomere but also non-sarcomere genes e.g., OBSCN was enriched for variants in adults, while VCL was more frequently mutated in children. It also helped us identify deleterious structural variants that would not readily be detected by panel testing or exome sequencing. These findings advance our understanding of the genetics of cardiomyopathy and have implications for using genetic information to guide predictions for age-related disease penetrance.
We included genes associated with cardiomyopathy that are also captured on one or more commercial gene panels. It is important when doing burden analyses to be inclusive since genes with lower penetrance may play a role in the penetrance of late-onset cardiomyopathy. Overall, we found a higher yield of deleterious variants in pediatric HCM patients (34.5%) compared to adult HCM patients (23.1%) especially for variants in Tier 1 genes, consistent with a recent report by Marston et al. (8). We also found a higher burden of de novo variants in cardiomyopathy genes in childhood compared to adult-onset cardiomyopathy. This information could be useful in pre-test counseling regarding diagnostic yield of genetic testing in patients by age of disease onset in the proband. Variants in pediatric patients clustered within more highly constrained coding regions of cardiomyopathy genes compared with those identified in adult patients. This was particularly true of variants in Tier 1 cardiomyopathy genes, including sarcomere genes. This is not entirely unexpected since variants in highly conserved regions are more likely to be intolerant of variation and therefore may manifest earlier penetrance compared to variants in less conserved regions that may show delayed penetrance since they may require an interaction with environmental factors and co-morbidities that usually accumulate with age [32]. The location of variants can also be an important predictor of penetrance as previously reported [33]. Deleterious variants in MYH7 in childhood HCM cases clustered in the myosin head and neck regions with very few variants seen in the tail domain unlike adult cases. The head and neck regions of MYH7 contain important functional domains i.e., ATPase site, actin binding site, the converter, and essential light chain, that play critical roles in myocardial contraction and energetics, and therefore variants in these regions are likely to cause more severe functional disturbances in encoded protein compared to variants in the tail domain [34,35,36].
At a gene level, MYH7 was the most affected gene in pediatric cardiomyopathy affecting 8.7% of cases (compared to 2.6% in adult HCM), while OBSCN was the most affected gene in adult HCM affecting 5.9% of adult cases (compared to 2.3% of pediatric HCM cases). A lower frequency of MYH7 variants in adults compared to the pediatric cohort or compared to findings in the SHaRE cohort may be related in part to differences in race and ethnicity (58% of patients in our study were of European ancestry compared to ~ 90% in SHaRe) [9], or unaccounted for bias towards recruiting genotype-elusive patients in the UK cohort.
Our study further confirmed that truncating variants in the A-band of titin remain the most common genetic cause of adult DCM accounting for 12% of adult cases as opposed to only 4.6% of pediatric DCM cases [29,30,31, 37]. Recent studies suggest that TTN variants may also act as genetic modifiers that predispose to heart failure in other conditions like myocarditis [38, 39]. Interestingly, OBSCN emerged as the second most mutated gene in adult DCM accounting for 7.3% of adult cases consistent with a recent report [40], but only 3.4% of pediatric DCM. Similar to TTN which is a giant protein, OBSCN is a giant protein in the sarcomere that interacts with the z-disc region of titin. Protein truncating variants in OBSCN were enriched in our cases compared to the gnomAD reference population, primarily in adults, reinforcing its role as a cardiomyopathy associated gene. This is consistent with findings from Wu et al. who showed a significant enrichment of OBSCN truncating variants in adult HCM compared to controls [40]. Whole genome sequencing also identified additional deleterious structural variants in OBSCN (and TTN) that have not been previously reported. At this time, OBSCN is considered a gene with limited evidence for association with cardiomyopathy [24, 25] probably due to variable or late penetrance. While most patients in our cohort harboring OBSCN variants were genotype-elusive for other Tier 1 genes, it is possible that OBSCN variants may act in some patients as a contributing or sensitizing variant as reported previously where OBSCN variants often occurred in conjunction with a second disease-associated variant [41]. This may account for the delayed penetrance observed in patients with only OBSCN variants. OBSCN missense and frameshift variants have been shown to co-segregate with HCM, DCM, and LVNC [40,41,42,43]. OBSCN deleterious variants in pediatric patients clustered around the protein kinase domains that interact with CDH2 known to cause ACM [44]. This provides a target for future research towards understanding the mechanism of effect of OBSCN variants. Finally, LoF variants in VCL, an important cytoskeletal protein with reported association with DCM [45, 46] were only seen in children, not in adults. This is consistent with a previous study that reported VCL LoF variants in pediatric DCM [47]. The patients harboring VCL variants in our cohort did not harbor any other pathogenic or likely pathogenic variants. Our findings add to the growing evidence for VCL LoF as a cause of childhood-onset DCM.
Sex-related phenotype differences in disease frequency and severity have been reported in cardiomyopathy with usually a higher prevalence, earlier onset and greater disease severity in male patients [12,13,14,15]. Other studies have reported conflicting findings [16,17,18]. In a study of HCM patients, female patients were more likely to harbor pathogenic or likely pathogenic variants in sarcomere genes compared to males [48]. In the study by van Velzen et al., there was no difference in genetic yield by sex but there was a lower genetic yield in women over 70 years of age at which age environmental factors may play a larger role than genetic factors [18]. The mean age of female patients in our cohort was 36 years. The higher yield of genetic testing in female patients in our cohort may reflect a cohort enriched for primary genetic cardiomyopathy. Variant CCR scores were also higher in female patients compared to male patients. Additionally, our study showed that z-disc genes were more frequently mutated in female DCM patients. Sex-related differences in genetics may explain some of the differences in clinical presentation and outcomes in cardiomyopathy. However, cardiac sex disparities may also be related to environmental differences or mechanisms regulated by sex hormones, and sex chromosomes [49].
Our findings of genetic differences between childhood and adult-onset cardiomyopathy have potential clinical implications. Children with cardiomyopathy had a higher diagnostic yield for variants in cardiomyopathy genes on sequencing compared to adults. This information can be used to inform pre-test counseling by providing greater precision in communicating the pre-test probability of a positive genetic test result based on age of the affected individual. It can help patients and families in decision making regarding genetic testing. Genetic results in cardiomyopathy patients are widely used for screening asymptomatic family members to identify at-risk individuals. However, it has been difficult to anticipate when clinical disease may manifest in an at-risk family member. While other genomic and environmental factors likely influence disease penetrance, our findings suggest that the genetic cause itself can be an important predictor of age of disease onset. A better understanding of how genetics guides disease penetrance may help in predictive counseling of family members, and in the approach to echocardiographic screening of at-risk family members.
The study has a few limitations. We limited our analysis to coding variants in known cardiomyopathy genes and did not include regulatory non-coding variants, in order to be consistent with what is offered in real world clinical gene panels. We adjusted for differences in race/ethnicity between cohorts but could not eliminate unaccounted for recruitment bias that may have contributed to some of the observed genetic differences. For example, although prior clinical genetic testing was not an exclusion criterion for patient eligibility, the Genomics England cohort may have been biased towards recruiting patients who were negative on previous panel testing. However, since the characteristics of patients not recruited is not available, it is not possible to ascertain if or to what extent there was a recruitment bias. Detailed clinical outcomes were not available in the adult cohort, therefore analysis of association of genotype with outcomes could not be performed. The role of environmental factors, co-morbidities and hormonal influences on age of onset or sex differences could also not be ascertained. Patients with clinically or genetically confirmed syndromes were excluded. Nonetheless, we found RASopathy associated variants in some pediatric and adult HCM patients on whole genome sequencing. We were able to do reverse phenotyping in accessible pediatric patients and found features suggestive of RASopathy in one patient. We did not exclude this patient post-hoc since the diagnosis was made later based on our genetic findings. We were not able to do reverse phenotyping in the UK patients to which we did not have clinical access.
Overall, our study identified a higher variant burden in pediatric cases, especially pediatric HCM compared to adults. The higher genomic constraints on variants found in pediatric patients suggest greater deleterious effects of these variants as a potential mechanism for earlier disease penetrance. Overall, our findings suggest that affected gene, variant location within the gene, and variant constraint scores may be useful in predicting disease penetrance by age and sex. This knowledge may inform pre-test counseling regarding diagnostic yield of genetic testing by patient age and help in predictive counseling of affected families by identifying individuals likely to develop early onset disease.
Abbreviations
- ACM:
-
Arrhythmogenic cardiomyopathy
- CCR:
-
Constrained coding region
- DCM:
-
Dilated cardiomyopathy
- gnomAD:
-
Genome Aggregation Database
- HCM:
-
Hypertrophic cardiomyopathy
- KS:
-
Kolmogorov-Smirnov
- LoF:
-
Loss-of-function
- LVNC:
-
Left ventricular non-compaction
- OR:
-
Odds Ratio
- RCM:
-
Restrictive cardiomyopathy
References
Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, Chung WK, Jefferies JL, Rossano JW, Castleberry CD, et al. Pediatric Cardiomyopathies. Circ Res. 2017;121:855–73. https://doi.org/10.1161/circresaha.116.309386.
Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, Hsu DT, Lin KY, Price JF, Wilkinson JD, et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation. 2019;140:e9–68. https://doi.org/10.1161/cir.0000000000000682.
Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, Parikh VN, Prakash S, Semsarian C, Sturm AC. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ Genom Precis Med. 2020;13:e000067. https://doi.org/10.1161/hcg.0000000000000067.
Tayal U, Ware JS, Lakdawala NK, Heymans S, Prasad SK. Understanding the genetics of adult-onset dilated cardiomyopathy: what a clinician needs to know. Eur Heart J. 2021;42:2384–96. https://doi.org/10.1093/eurheartj/ehab286.
Ware SM, Wilkinson JD, Tariq M, Schubert JA, Sridhar A, Colan SD, Shi L, Canter CE, Hsu DT, Webber SA, et al. Genetic Causes of Cardiomyopathy in Children: First Results From the Pediatric Cardiomyopathy Genes Study. J Am Heart Assoc. 2021;10:e017731. https://doi.org/10.1161/jaha.120.017731.
Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J Am Coll Cardiol. 2016;68:2871–86. https://doi.org/10.1016/j.jacc.2016.08.079.
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–16. https://doi.org/10.1161/circulationaha.106.174287.
Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138:1387–98. https://doi.org/10.1161/circulationaha.117.033200.
Marston NA, Han L, Olivotto I, Day SM, Ashley EA, Michels M, Pereira AC, Ingles J, Semsarian C, Jacoby D, et al. Clinical characteristics and outcomes in childhood-onset hypertrophic cardiomyopathy. Eur Heart J. 2021;42:1988–96. https://doi.org/10.1093/eurheartj/ehab148.
Patel MD, Mohan J, Schneider C, Bajpai G, Purevjav E, Canter CE, Towbin J, Bredemeyer A, Lavine KJ. Pediatric and adult dilated cardiomyopathy represent distinct pathological entities. JCI Insight. 2017;2. https://doi.org/10.1172/jci.insight.94382
Wittlieb-Weber CA, Lin KY, Zaoutis TE, O’Connor MJ, Gerald K, Paridon SM, Shaddy RE, Rossano JW. Pediatric versus adult cardiomyopathy and heart failure-related hospitalizations: a value-based analysis. J Cardiac Fail. 2015;21:76–82. https://doi.org/10.1016/j.cardfail.2014.10.011.
Cannatà A, Fabris E, Merlo M, Artico J, Gentile P, Pio Loco C, Ballaben A, Ramani F, Barbati G, Sinagra G. Sex Differences in the Long-term Prognosis of Dilated Cardiomyopathy. Can J Cardiol. 2020;36:37–44. https://doi.org/10.1016/j.cjca.2019.05.031.
Olivotto I, Maron MS, Adabag AS, Casey SA, Vargiu D, Link MS, Udelson JE, Cecchi F, Maron BJ. Gender-related differences in the clinical presentation and outcome of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;46:480–7. https://doi.org/10.1016/j.jacc.2005.04.043.
Rowin EJ, Maron MS, Wells S, Patel PP, Koethe BC, Maron BJ. Impact of Sex on Clinical Course and Survival in the Contemporary Treatment Era for Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2019;8:e012041. https://doi.org/10.1161/jaha.119.012041.
Siontis KC, Ommen SR, Geske JB. Sex, Survival, and Cardiomyopathy: Differences Between Men and Women With Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2019;8:e014448. https://doi.org/10.1161/jaha.119.014448.
Alimohamed MZ, Johansson LF, Posafalvi A, Boven LG, van Dijk KK, Walters L, Vos YJ, Westers H, Hoedemaekers YM, Sinke RJ, et al. Diagnostic yield of targeted next generation sequencing in 2002 Dutch cardiomyopathy patients. Int J Cardiol. 2021;332:99–104. https://doi.org/10.1016/j.ijcard.2021.02.069.
Meyer S, van der Meer P, van Tintelen JP, van den Berg MP. Sex differences in cardiomyopathies. Eur J Heart Fail. 2014;16:238–47. https://doi.org/10.1002/ejhf.15.
van Velzen HG, Schinkel AFL, Baart SJ, Huurman R, van Slegtenhorst MA, Kardys I, Michels M. Effect of Gender and Genetic Mutations on Outcomes in Patients With Hypertrophic Cardiomyopathy. Am J Cardiol. 2018;122:1947–54. https://doi.org/10.1016/j.amjcard.2018.08.040.
Fung A, Manlhiot C, Naik S, Rosenberg H, Smythe J, Lougheed J, Mondal T, Chitayat D, McCrindle BW, Mital S. Impact of prenatal risk factors on congenital heart disease in the current era. J Am Heart Assoc. 2013;2:e000064. https://doi.org/10.1161/jaha.113.000064.
Papaz T, Liston E, Zahavich L, Stavropoulos DJ, Jobling RK, Kim RH, Reuter M, Miron A, Oechslin E, Mondal T, et al. Return of genetic and genomic research findings: experience of a pediatric biorepository. BMC Med Genomics. 2019;12:173. https://doi.org/10.1186/s12920-019-0618-0.
Papaz T, Safi M, Manickaraj AK, Ogaki C, BreatonKyryliuk J, Burrill L, Dodge C, Chant-Gambacort C, Walter LL, Rosenberg H, et al. Factors influencing participation in a population-based biorepository for childhood heart disease. Pediatrics. 2012;130:e1198-1205. https://doi.org/10.1542/peds.2012-0687.
Smedley D, Smith KR, Martin A, Thomas EA, McDonagh EM, Cipriani V, Ellingford JM, Arno G, Tucci A, Vandrovcova J, et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care - Preliminary Report. N Engl J Med. 2021;385:1868–80. https://doi.org/10.1056/NEJMoa2035790.
Lesurf R, Said A, Akinrinade O, Breckpot J, Delfosse K, Liu T, Yao R, Persad G, McKenna F, Noche RR, et al. Whole genome sequencing delineates regulatory, copy number, and cryptic splice variants in early onset cardiomyopathy. NPJ Genom Med. 2022;7:18. https://doi.org/10.1038/s41525-022-00288-y.
Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, Dougherty K, Harrison SM, McGlaughon J, Milko LV, et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ Genom Precis Med. 2019;12:e002460. https://doi.org/10.1161/circgen.119.002460.
Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, Celeghin R, Edwards M, Fan J, Ingles J, et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation. 2021;144:7–19. https://doi.org/10.1161/circulationaha.120.053033.
Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, Mazaika E, de Marvao A, Dawes TJW, et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation. 2020;141:387–98. https://doi.org/10.1161/circulationaha.119.037661.
Thomson KL, Ormondroyd E, Harper AR, Dent T, McGuire K, Baksi J, Blair E, Brennan P, Buchan R, Bueser T, et al. Analysis of 51 proposed hypertrophic cardiomyopathy genes from genome sequencing data in sarcomere negative cases has negligible diagnostic yield. Genet Med. 2019;21:1576–84. https://doi.org/10.1038/s41436-018-0375-z.
Walsh R, Buchan R, Wilk A, John S, Felkin LE, Thomson KL, Chiaw TH, Loong CCW, Pua CJ, Raphael C, et al. Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur Heart J. 2017;38:3461–8. https://doi.org/10.1093/eurheartj/ehw603.
Akinrinade O, Alastalo TP, Koskenvuo JW. Relevance of truncating titin mutations in dilated cardiomyopathy. Clin Genet. 2016;90:49–54. https://doi.org/10.1111/cge.12741.
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28. https://doi.org/10.1056/NEJMoa1110186.
Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra276. https://doi.org/10.1126/scitranslmed.3010134.
Tadros R, Francis C, Xu X, Vermeer AMC, Harper AR, Huurman R, KeluBisabu K, Walsh R, Hoorntje ET, Te Rijdt WP, et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet. 2021;53:128–34. https://doi.org/10.1038/s41588-020-00762-2.
Homburger JR, Green EM, Caleshu C, Sunitha MS, Taylor RE, Ruppel KM, Metpally RP, Colan SD, Michels M, Day SM, et al. Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc Natl Acad Sci USA. 2016;113:6701–6. https://doi.org/10.1073/pnas.1606950113.
Debold EP, Schmitt JP, Patlak JB, Beck SE, Moore JR, Seidman JG, Seidman C, Warshaw DM. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol. 2007;293:H284-291. https://doi.org/10.1152/ajpheart.00128.2007.
Spudich JA, Aksel T, Bartholomew SR, Nag S, Kawana M, Yu EC, Sarkar SS, Sung J, Sommese RF, Sutton S, et al. Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human β-cardiac myosin. J Exp Biol. 2016;219:161–7. https://doi.org/10.1242/jeb.125930.
Toepfer CN, Garfinkel AC, Venturini G, Wakimoto H, Repetti G, Alamo L, Sharma A, Agarwal R, Ewoldt JF, Cloonan P, et al. Myosin Sequestration Regulates Sarcomere Function, Cardiomyocyte Energetics, and Metabolism, Informing the Pathogenesis of Hypertrophic Cardiomyopathy. Circulation. 2020;141:828–42. https://doi.org/10.1161/circulationaha.119.042339.
Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–8. https://doi.org/10.1038/gim.2013.204.
Seidel F, Holtgrewe M, Al-Wakeel-Marquard N, Opgen-Rhein B, Dartsch J, Herbst C, Beule D, Pickardt T, Klingel K, Messroghli D, et al. Pathogenic Variants Associated With Dilated Cardiomyopathy Predict Outcome in Pediatric Myocarditis. Circ Genom Precis Med. 2021;14:e003250. https://doi.org/10.1161/CIRCGEN.120.003250.
Kontorovich AR, Patel N, Moscati A, Richter F, Peter I, Purevjav E, Selejan SR, Kindermann I, Towbin JA, Bohm M, et al. Myopathic Cardiac Genotypes Increase Risk for Myocarditis. JACC Basic Transl Sci. 2021;6:584–92. https://doi.org/10.1016/j.jacbts.2021.06.001.
Wu G, Liu J, Liu M, Huang Q, Ruan J, Zhang C, Wang D, Sun X, Jiang W, Kang L, et al. Truncating Variants in OBSCN Gene Associated With Disease-Onset and Outcomes of Hypertrophic Cardiomyopathy. Circ Genom Precis Med. 2021;14:e003401. https://doi.org/10.1161/circgen.121.003401.
Marston S. Obscurin variants and inherited cardiomyopathies. Biophys Rev. 2017;9:239–43. https://doi.org/10.1007/s12551-017-0264-8.
Grogan A, Kontrogianni-Konstantopoulos A. Unraveling obscurins in heart disease. Pflugers Arch. 2019;471:735–743. https://doi.org/10.1007/s00424-018-2191-3
Hu LR, Kontrogianni-Konstantopoulos A. Proteomic Analysis of Myocardia Containing the Obscurin R4344Q Mutation Linked to Hypertrophic Cardiomyopathy. Front Physiol. 2020;11:478. https://doi.org/10.3389/fphys.2020.00478.
Mayosi BM, Fish M, Shaboodien G, Mastantuono E, Kraus S, Wieland T, Kotta MC, Chin A, Laing N, Ntusi NB, et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ Cardiovasc Genet. 2017;10. https://doi.org/10.1161/circgenetics.116.001605.
Sarker M, Lee HT, Mei L, Krokhotin A, de Los Reyes SE, Yen L, Costantini LM, Griffith J, Dokholyan NV, Alushin GM, et al. Cardiomyopathy Mutations in Metavinculin Disrupt Regulation of Vinculin-Induced F-Actin Assemblies. J Mol Biol. 2019;431:1604–18. https://doi.org/10.1016/j.jmb.2019.02.024.
Zemljic-Harpf AE, Miller JC, Henderson SA, Wright AT, Manso AM, Elsherif L, Dalton ND, Thor AK, Perkins GA, McCulloch AD, et al. Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol Cell Biol. 2007;27:7522–37. https://doi.org/10.1128/mcb.00728-07.
Hawley MH, Almontashiri N, Biesecker LG, Berger N, Chung WK, Garcia J, Grebe TA, Kelly MA, Lebo MS, Macaya D, et al. An assessment of the role of vinculin loss of function variants in inherited cardiomyopathy. Hum Mutat. 2020;41:1577–87. https://doi.org/10.1002/humu.24061.
Lakdawala NK, Olivotto I, Day SM, Han L, Ashley EA, Michels M, Ingles J, Semsarian C, Jacoby D, Jefferies JL, et al. Associations Between Female Sex, Sarcomere Variants, and Clinical Outcomes in Hypertrophic Cardiomyopathy. Circ Genom Precis Med. 2021;14:e003062. https://doi.org/10.1161/circgen.120.003062.
Shi W, Sheng X, Dorr KM, Hutton JE, Emerson JI, Davies HA, Andrade TD, Wasson LK, Greco TM, Hashimoto Y, et al. Cardiac proteomics reveals sex chromosome-dependent differences between males and females that arise prior to gonad formation. Dev Cell. 2021;56:3019-3034.e3017. https://doi.org/10.1016/j.devcel.2021.09.022.
Acknowledgements
We acknowledge the Labatt Family Heart Centre Biobank at the Hospital for Sick Children for access to DNA samples for whole genome sequencing, and The Centre for Applied Genomics at the Hospital for Sick Children for performing whole genome sequencing. This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. We acknowledge the Genomics England Research Consortium members: Ambrose, J. C.1; Arumugam, P.1; Bleda, M.1; Boardman-Pretty, F.1,2; Boustred, C. R.1; Brittain, H.1; Caulfield, M. J.1,2; Chan, G. C.1; Fowler, T.1; Giess A.1; Hamblin, A.1; Henderson, S.1,2; Hubbard, T. J. P.1; Jackson, R.1; Jones, L. J.1,2; Kasperaviciute, D.1,2; Kayikci, M.1; Kousathanas, A.1; Lahnstein, L.1; Leigh, S. E. A.1; Leong, I. U. S.1; Lopez, F. J.1; Maleady-Crowe, F.1; Moutsianas, L.1,2; Mueller, M.1,2; Murugaesu, N.1; Need, A. C.1,2; O‘Donovan P.1; Odhams, C. A.1; Patch, C.1,2; Perez-Gil, D.1; Pereira, M.B.1; Pullinger, J.1; Rahim, T.1; Rendon, A.1; Rogers, T.1; Savage, K.1; Sawant, K.1; Scott, R. H.1; Siddiq, A.1; Sieghart, A.1; Smith, S. C.1; Sosinsky, A.1,2; Stuckey, A.1; Tanguy, M.1; Thomas, E. R. A.1,2; Thompson, S. R.1; Tucci, A.1,2; Walsh, E.1; Welland, M. J.1; Williams, E.1; Witkowska, K.1,2; Wood, S. M.1,2.
1. Genomics England, London, UK
2. William Harvey Research Institute, Queen Mary University of London, London, EC1M 6BQ, UK.
Funding
We acknowledge the funders of our work—Ted Rogers Centre for Heart Research (SM), Data Sciences Institute at the University of Toronto (SM), the Canadian Institutes of Health Research (PJT 175034) (SM), and the Canadian Institutes of Health Research (ENP 161429), under the frame of ERA PerMed (SM, RL). SM holds the Heart and Stroke Foundation of Canada / Robert M Freedom Chair in Cardiovascular Science. EO holds the Bitove Family Professorship of Adult Congenital Heart Disease. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care) and funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure.
Author information
Authors and Affiliations
Consortia
Contributions
OA and SM conceived and designed the work, drafted the work and substantively revised it. SM acquired funding for the project. All authors contributed to acquisition, analysis or interpretation of data. All authors approved the submitted version and have agreed both to be personally accountable for the author's own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature.
Corresponding author
Ethics declarations
Conflicts of Interest
SM is on the Advisory Board of Bristol Myers Squibb, and Tenaya Therapeutics. The remaining authors have nothing to disclose.
Human Subjects Statement
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Animal Studies
No animal studies were carried out by the authors for this article.
Data Statement
Sequencing data from the Ontario Registry are deposited in the European Genome-Phenome Archive (EGA) under accession EGAS00001004929, and are available for download upon approval by the Data Access Committee. The 100,000 Genomes Project data are available to researchers and trainees using the Genomics England Research Environment upon institutional approval through their Participation Agreement process. Additional data generated or analyzed during this study are included in the supplementary information files, and additional raw data used for figures and results are available from the corresponding author on reasonable request.
Author Statement
All authors contributed to the study conception and design, and to data collection. Data analysis was performed by [Oyediran Akinrinade] [Robert Lesurf]. The first draft of the manuscript was written by [Oyediran Akinrinade]. Funding was acquired by [Seema Mital]. All authors commented on manuscript versions and read and approved the final manuscript.
Additional information
Associate Editor Roddy Walsh oversaw the review of this article
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Akinrinade, O., Lesurf, R., Genomics England Research Consortium. et al. Age and Sex Differences in the Genetics of Cardiomyopathy. J. of Cardiovasc. Trans. Res. 16, 1287–1302 (2023). https://doi.org/10.1007/s12265-023-10411-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-023-10411-8