
Abstract
Mechanosensation (the ultimate conversion of a mechanical stimulus into a biochemical signal) as well as mechanotransduction (transmission of mechanically induced signals) belong to the most fundamental processes in biology. These effects, because of their dynamic nature, are particularly important for the cardiovascular system. Therefore, it is not surprising that defects in cardiac mechanosensation, are associated with various types of cardiomyopathy and heart failure. However, our current knowledge regarding the genetic basis of impaired mechanosensation in the cardiovascular system is beginning to shed light on this subject and is at the centre of this brief review.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease (CVD) is the largest source of morbidity and mortality in the European Union; the financial burden totals 169 billion Euros annually with healthcare accounting for 62% of costs [1] and the WHO expects CVD to become the leading cause of death worldwide by 2015. In addition to the economic impact, individuals affected by CVD experience a significantly impaired quality of life as well as a reduced life expectancy (i.e. 50% of all patients affected by severe heart failure die within 5 years of diagnosis, which makes the mortality rate for heart failure comparable to that of cancer).
Unfortunately, and in contrast to the socioeconomic importance of CVD, our present knowledge regarding the underlying disease causing molecular mechanisms of heart failure, remains elusive as the foundation for novel therapeutic approaches are limited.
Cardiomyopathies are primary diseases of the heart muscle and major causes of heart failure associated with high morbidity and mortality [2]. In particular, hypertrophic cardiomyopathy (HCM; prevalence, 1/500) and dilated cardiomyopathy (DCM; prevalence, 1/2,500) are very common. Cardiomyopathies can also be due to a variety of different infections (“myocarditis”), the consequence of exposition to many different toxins or drugs, including alcohol and some rare cases due to mutations in the mitochondrial genome. However, an estimated 80% of all HCM and probably at least 30% of all DCM cases are hereditary, which points to the importance of genetic factors [3, 4]. In the last 10–20 years, a variety of different mutations in a wide range of different genes encoding mainly cytoskeletal or sarcomeric components have been identified as causative in several familial cardiomyopathies (for a recent review please see [5]). The analysis of these genes, including the analysis of their mutations is not only important for CVD per se, but this research offers the unique possibility to use this “experiment of nature” to get a deeper insight into gene function and cellular biology, particularly because our knowledge of how single basepair changes cause complex diseases is very limited.
As such, a variety of different mechanisms have been proposed to explain how changes in the genetic composition are associated with cardiomyopathy and different forms of heart failure. Among them are defects in force generation and or force transmission, changes in calcium sensitivity and metabolism, in mechanosensation as well as in mechanotransduction. Cardiomyopathy and heart failure due to imperfect mechanosensation is a relatively new hypothesis to explain the pathogenesis of cardiac diseases. In addition to this, defects in mechanosensation might be difficult to prove in the experimental context because large-scale assays to check for such defects are missing.
Here, we discuss briefly some of the most important mechanisms involved in cardiac mechanosensation as well as their implications for different types of cardiomypathy and human heart failure. We focus primarily on cytoskeletal and sarcomeric components rather than on ion channels, which have been recently elegantly reviewed [6].
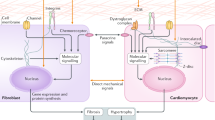
Life as we know it today is unimaginable without mechanosensation—a highly conserved process found in a wide variety of different cell types, tissues, organs and even at the level of single-cell organisms [7]. Different environmental conditions during evolution led to the development of extremely divergent stretch sensing mechanisms in these cell types. As a consequence, there is no common “mechanosensation mechanism” present nor is there “one single mechanosensation mechanism” responsible for all mechanosensory effects observable in a single cell. This is particularly true for cardiomyocytes where different mechanisms are simultaneously involved to integrate a multitude of mechanical stimuli into biochemical signals. These stimuli include passive effects induced by blood flow and shear stress as well as active effects caused by cycles of contraction and relaxation of single cardiomyocytes. From this point of view, it is also clear that cardiomyocytes are in need of different types of mechanosensors such as length and force sensors that are necessary to identify different mechanical stimuli (Fig. 1).

The figure summarises some of the postulated mechanosensory pathways active in cardiomyocytes, namely: (1) AT1 receptor-mediated pathway, (2) integrin-mediated pathway, (3) Z-disc-mediated pathway, (4) GPCR/N2B/FHL1/Erk 2 pathway, (5) MARP-mediated pathway, (6) titin kinase-mediated pathway. Interestingly, pathways 3–6 are sarcomere related. Abbreviations: AT1 angiotensin II receptor, ILK integrin-linked kinase, FAK focal adhesion kinase, GPCR G protein-coupled receptor, SRF serum response factor, MARP muscle ankyrin repeat protein
Length (Strain) Sensor—Titin
Titin is the largest molecule known in biology, it may add up to 4.2 mDa and has an in situ length of about 1–2 μm in the passive cardiac myocyte, spanning from the Z-disc to the M-band of the sarcomere. Active force in the form of stretch is related to the change in the length of sarcomeres within the ventricular myocytes. When a cardiac myocyte is stretched, there is an increase from resting sarcomere length (SL) approximately 1.9 μm up to a maximum at a length (L max) of =2.2–2.3 μm (please see for an review on this topic the article by Granzier and Labeit [8]). Beside the thin (actin) and thick filaments (myosin), titin is called the “third” filament. As such, it is no surprise that this molecule fulfils many different functions, such as serving as a molecular ruler. Titin is also an important factor during embryonic development and influences via its interactions with myosin binding protein C directly myocardial contractile function [9]. However, one of the most important functions of this “titanic” molecule is to retain the cardiomyocyte in an optimal length, which is achieved in the myocardium by three different molecular springs: the PEVK (rich in proline, glutamate, valine and lysine), the tandem immunoglobulin (Ig) as well as the N2B domains, which encode cardiac specific-titin transcripts. All three domains are localised in the I-band of the sarcomere perfectly suited to sense any change in sarcomeric length (please see for a review on titin elastic domains the article by Granzier and Labeit [10])
The PEVK, tandem Ig and N2B domains interact with a variety of different proteins, for example the N2-A domain binds (next to a calpain protease, p94 or calpain-3 binding site) a protein complex consisting of muscle ankyrin repeat proteins (MARPs, including diabetes-associated ankyrin repeat proteins (DARP), cardiac ankyrin repeat proteins (CARP) and Ankrd-2), which in turn bind myopalladin. Upon stretch, DARP and CARP translocate to the nucleus where they probably participate in the orchestration of a stretch-dependent gene programme [11]. This is supported by the fact that Ankrd-1 and 2 bind transcription factors, such as PML, YB-1 or p53, and are able to potentiate the effects of p53 on the p21 promoter [12, 13]. Interestingly, ablation of the PEVK domain in a genetically altered mouse model leads to a defect in passive elasticity, myocardial hypertrophy and diastolic heart failure, together with FHL2 upregulation [14].
Another link between titin, length sensation and gene expression is provided by the binding of N2B titin to four and a half LIM domain protein 1 (FHL1), Raf, MEK1/2 and ERK2 which links titin directly to G protein-coupled (GPCR) signalling and regulates cardiomyocyte length and hypertrophic gene expression [15].
Over the last decade, a number of titin mutations associated with various muscle pathologies such as DCM, HCM or tibial muscular dystrophy have been reported but the precise series of molecular events leading to the phenotypes remain unclear (for an overview please see [16]). It is impossible to discuss all of these genetic variants here, however mutations particularly localised in the N2B region such as the HCM-associated S3799T, which increases binding to FHL2 and the DCM-associated Q4053ter mutation which decreases titin's affinity to FHL2 do probably have effects on mechanosensation. Another mutation, the S4465D-titin, is directly localised in the FHL2 binding site [17, 18].
Another link to mechanosensation is probably provided by mutations localised in ANKRD1 or CARP, where three mutations have been reported recently in various patients affected by DCM. In particular, the P105S and M184I substitutions result in loss of binding with talin, a key binding partner of the beta-integrin subunit of the integrin complex and as such links CARP not only via titin but also via talin potentially to mechanosensation. In addition, the M184I variant results also in loss of CARP interaction with FHL2 [19]. Interestingly, the same group identified three additional CARP mutations (P52A, T123M, I280V), this time associated with HCM leading to an increase of affinity between CARP and titin [20].
Vice versa, the recently discovered G48S—FHL2 variant abolishes to a large extent the titin/FHL2 interaction and has been found in an individual affected by DCM [21]. However, although the data provided do strongly support the idea that human mutations affect titin-mediated mechanosensation and lead to cardiomyopathy and heart failure, it remains to be seen if more detailed and additional binding studies in solution can confirm this concept.
Moreover, the elastic properties of the N2-B domain of titin are significantly influenced by phosphorylation which can in principle affect the mechanosensitive function of this segment—however, experimental prove for this hypothesis is missing [16]. It can be hypothesised that mutations affecting these phosphorylation sites may also affect titin's ability to sense stress.
A completely different mechanism of length sensation has been proposed for titin's kinase domain, located at the M-band of the sarcomere. This domain has been found to interact with signalling molecules such as Nbr1, p62 and the E3 ubiquitin ligase, MURF2 [22]. Mechanical inactivity is associated with nuclear translocation of MURF2 in exchange for serum response factor (SRF) and transcriptional repression. Interestingly, a titin kinase mutation (R279W, located in the αR1 part of the regulatory tail) is associated with hereditary myopathy with early respiratory failure [22]. Titin kinase might be involved in short-term mechanosensing, e.g. by phosphorylating a target protein depending on the sarcomeric stretch state [23], but also in long-term effects such as changing the hypertrophic gene expression pattern. However, a titin kinase mutation linked to human cardiomyopathy has not yet been reported. Furthermore, in a recent report where MURF1 and MURF2 deficient animals were employed, it turned out that the relationship between MURF2 and SRF is probably even more complex [24]. Therefore it is necessary that future studies are carried out, including the generation of conditional knock-in models for this titin kinase mutation followed by the detailed analysis of its distinct function in cardiac mechanosensation.
The Sarcomeric Z-Disc
Z-discs define the lateral boundaries of sarcomeres, the smallest functional units in cross-striated muscles. They belong to the most complex macromolecular structures in biology which are probably composed of hundreds of different proteins [25]. Because of their anatomical localization, they are nodal points for mechanosensation, mechanotransduction, for mechanical stability and force transmission as well as for embryonic development. They probably do not function as length sensors like titin but due to their exposition to stress particularly during systole, they may serve as strain (tension, force) sensors. As a consequence, almost all molecular components of the sarcomeric Z-disc might be involved in mechanosensation and mutations in their genes might give rise to cardiomyopathy and heart failure via defects in this system. In fact, the sarcomeric Z-disc is increasingly recognised as a hot spot for cardiomyopathy and heart failure causing mutations [26, 27].
One of the implicated genes in cardiomyopathy and heart failure is the muscle LIM protein (MLP) or cysteine rich protein 3. It is a 194 amino-acids protein that is predominantly expressed in muscle tissues, with an important role in myocardial function and performance (for a brief overview please see: [28]). This protein has been shown to be involved in cardiac mechanosensation, possibly by functioning as a tension sensor. MLP binds to telethonin (TCAP) which is known to interact with titin's extreme N-terminal Z1/Z2 domains. Upon an increase in biomechanical stress, MLP has been shown to translocate to the nucleus using its nuclear localization signal located within the first glycine rich domain (GRD) [29]. Interestingly, mutations in MLP as well as telethonin (TCAP) have been shown to be associated with different forms of cardiomyopathy [30–32]. Although MLP, telethonin and titin's N-terminal Z1/Z2 domain were suggested to build a macromolecular—mechanosensitive structure, the precise molecular mechanism by which this structure perceives mechanical stimuli and transforms them into biochemical signals remains unclear. Alternatively, Furukawa et al. have speculated the interaction of telethonin with titin and MinK, the β-subunit of potassium current (Iks) channel, may contribute to a stretch-dependent regulation of potassium flux in cardiac muscle, serving as a “mechano-electrical feedback” system [33]. In this context, it is probably important to mention our finding of an interaction between MLP and integrin-linked kinase (ILK) as well as of additional mutations localised in the ILK as well as laminin alpha 4 genes, which are also associated with cardiomyopathy and heart failure [34, 35]. Although we did not look for defects in mechanosensation when we performed this set of experiments, defects in this system seem to be likely because integrins are well known to be involved in the perception and transmission of mechanical stimuli (Table 1).
Intermediate Filaments
Intermediate filaments provide physical links between Z-discs of peripheral sarcomeres and the sarcolemma at costameres (and via these to the ECM). In addition to this, also between Z-discs of adjacent sarcomeres, Z-discs and nuclei and cell-to-cell contacts at the desmosomes. Desmin is a major intermediate filament and mainly present in extranuclear compartments, whereas lamin A/C is a nuclear membrane constituent. Both contain elastic domains, hence are able to react to changes in cell morphology, and respond by interacting with a wide variety of different proteins [36]. Mutations in desmin cause muscular dystrophy as well as cardiomyopathies associated with conduction blocks and arrhythmias leading to sudden death as well as arrhythmogenic right ventricular cardiomyopathy [37, 38]. Several mechanisms have been made accountable for the effects of these mutations, one of which is certainly that absence of desmin at the Z-disc impairs the generation and transmission of force through misalignment of sarcomere units [39]. Another mechanism may involve disruption of the physical pathway between the sarcomere and the nuclear membrane which is mainly provided by desmin and where mutations might cause disruption of the translation of changes in sarcomere length into changes in gene expression [40].
In addition, lamin A/C mutations are the most common cause of dilated cardiomyopathy and more than 200 different mutations are known in this gene, which are linked to more than ten different types of disease [36], among them cardiomyopathy, muscular dystrophy and lipodystrophy. The underlying disease causing molecular mechanisms for mutations located in this gene are less well understood, but studies based on desmin and lamin A/C knockout animal models [41] suggest mutations in these genes are linked to defects in mechanosensation and mechanotransduction [36]. Recent reports demonstrated the involvement of nesprin, another nuclear envelope protein, in the development of cardiomyopathy and heart failure in lamin A/C-deficient animals, which points to the importance of nuclear membrane stability in the myocardium. Moreover, lamin A/C-deficient animals have additional defects in the hypo-osmotic reaction, pointing to a possible defect in mechanosensation and perhaps defects in mechano-transcriptional coupling [42]. The same group found as well that carvedilol treatment leads to significant improvement of the cardiac phenotype in these animals—it might be possible that defects in mechanosensation are targetable by this strategy [43].
Angiotensin 2
Angiotensin 2 is a well-known mediator of cardiomyocyte hypertrophy. Stretch of cardiomyocytes is associated with the local release of angiotensin 2, activating the angiotensin 2 receptor 1 (AT1), which mediates mechanosensory effects, perhaps via activation of MAP kinase signalling [44].
Even more important is that the AT1 receptor, without binding angiotensin 2, is directly activated by stretch [45, 46]. This surprising finding demonstrates a previously unappreciated role of the AT1 receptor in mechanosensation and brings this molecule directly into the context of stress perception in single cells. It is conceptually novel that the AT1 receptor, a member of G protein-coupled receptor, is a mechanical force-transducing molecule and mediates mechanical stress-induced cellular responses. An increase in left ventricular mass which was produced by abdominal aortic constriction, without significant increase in plasma renin activity, was completely prevented with an ACE inhibitor or AngII type 1 (AT1) receptor antagonists without any change in afterload. In support of this finding, Frank et al. have reported that the AT1 receptor blocker irbesartan markedly blunted stretch-mediated GDF15 and Hmox1 upregulation, suggesting that the angiotensin receptor tranduces the biomechanical induction of these genes [47]. Besides this, AngII activates many protein kinases, such as ERKs, JNK, p38MAP kinase and JAK/STAT, and also induces expression of IEG such as c-fos and c-jun genes as well as increases protein synthesis in cardiac myocytes.
In summary, these results strongly suggest that the local renin–angiotensin system, but not the circulating one, is a major mediator of pressure overload-induced cardiac hypertrophy. Once mechanical stress is perceived and converted into biochemical signals, the activated signal transduction pathway or pathways leading to changes in cardiac myocyte structure and metabolism seem to be similar to those pathways known to be activated by various humoral factors such as growth factors, hormones and cytokines in many other cells.
Mechanosensation and Genetics—Outlook
In summary, the above described, novel length and tension-dependent intra- or extra-sarcomeric mechanosensation mechanisms may play a significant role in the regulation of myocardial hypertrophy, atrophy and sarcopenia. Moreover, mutations in these systems may lead to cardiomyopathy, heart failure and arrhythmogenesis. Studies related to physiological stretch signalling have been carried out decades ago, but these reports were the first to specifically identify human mutations in some of these components and to link them to disease [31, 32, 34].
Although MLP and telethonin as well as titin's kinase domain have been directly or indirectly implicated in the process of mechanosensation and human mutations have been linked to types of heart and or muscle pathologies, at present, most other mutations known to cause various types of cardiac pathology have not been studied under this aspect in such detail. One of the reasons as to why they have not been examined in this context is due to the complexity associated with the analysis of mechanosensation, which is a multifaceted response to various mechanical stimuli and as such requires the presence of a living cell or at least functioning large macromolecular protein compounds.
In addition, future research should be directed to translate our knowledge of mechanosensation into novel therapeutic strategies, possible targets might include FAK, ILK (kinase inhibitors, activators) and or interventions at the level of nuclear translocation of some of the mediators of mechanosensation (such as MLP, MARP).
Abbreviations
- CVD:
-
Cardiovascular disease
- DCM:
-
Dilated cardiomypathy
- FAK:
-
Focal adhesion kinase
- HCM:
-
Hypertrophic cardiomyopathy
- Ig:
-
Immunoglobulin
- ILK:
-
Integrin-linked kinase
- MARP:
-
Muscle ankyrin repeat protein
- SL:
-
Sarcomere length
References
Leal, J., Luengo-Fernandez, R., Gray, A., Petersen, S., & Rayner, M. (2006). Economic burden of cardiovascular diseases in the enlarged European Union. European Heart Journal, 27(13), 1610–1619.
Maron, B. J., Towbin, J. A., Thiene, G., Antzelevitch, C., Corrado, D., Arnett, D., et al. (2006). Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation, 113(14), 1807–1816.
Carballo, S., Robinson, P., Otway, R., Fatkin, D., Jongbloed, J. D., de Jonge, N., et al. (2009). Identification and functional characterization of cardiac troponin I as a novel disease gene in autosomal dominant dilated cardiomyopathy. Circulation Research, 105(4), 375–382. doi:10.1161/CIRCRESAHA.109.196055.
Watkins, H., Ashrafian, H., & McKenna, W. J. (2008). The genetics of hypertrophic cardiomyopathy: Teare redux. Heart, 94(10), 1264–1268. doi:10.1136/hrt.2008.154104.
Kimura, A. (2010). Molecular basis of hereditary cardiomyopathy: abnormalities in calcium sensitivity, stretch response, stress response and beyond. Journal of Human Genetics, 55(2), 81–90. doi:10.1038/jhg.2009.138.
Inoue, R., Jian, Z., & Kawarabayashi, Y. (2009). Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacology & Therapeutics, 123(3), 371–385. doi:10.1016/j.pharmthera.2009.05.009.
Kung, C. (2005). A possible unifying principle for mechanosensation. Nature, 436(7051), 647–654. doi:10.1038/nature03896.
Granzier, H., & Labeit, S. (2002). Cardiac titin: an adjustable multi-functional spring. Journal de Physiologie, 541(Pt 2), 335–342.
Linke, W. A. (2008). Sense and stretchability: The role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovascular Research, 77(4), 637–648.
Granzier, H. L., & Labeit, S. (2004). The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circulation Research, 94(3), 284–295. doi:10.1161/01.RES.0000117769.88862.F8.
Miller, M. K., Bang, M. L., Witt, C. C., Labeit, D., Trombitas, C., Watanabe, K., et al. (2003). The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. Journal of Molecular Biology, 333(5), 951–964.
Kojic, S., Medeot, E., Guccione, E., Krmac, H., Zara, I., Martinelli, V., et al. (2004). The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. Journal of Molecular Biology, 339(2), 313–325.
Kojic, S., Nestorovic, A., Rakicevic, L., Belgrano, A., Stankovic, M., Divac, A., et al. (2010). A novel role for cardiac ankyrin repeat protein Ankrd1/CARP as a co-activator of the p53 tumor suppressor protein. Archives of Biochemistry and Biophysics, 502(1), 60–67. doi:10.1016/j.abb.2010.06.029.
Granzier, H. L., Radke, M. H., Peng, J., Westermann, D., Nelson, O. L., Rost, K., et al. (2009). Truncation of titin's elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circulation Research, 105(6), 557–564. doi:10.1161/CIRCRESAHA.109.200964.
Sheikh, F., Raskin, A., Chu, P. H., Lange, S., Domenighetti, A. A., Zheng, M., et al. (2008). An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. Journal of Clinical Investigation, 118(12), 3870–3880.
Kruger, M., & Linke, W. A. (2009). Titin-based mechanical signalling in normal and failing myocardium. Journal of Molecular and Cellular Cardiology, 46(4), 490–498.
Itoh-Satoh, M., Hayashi, T., Nishi, H., Koga, Y., Arimura, T., Koyanagi, T., et al. (2002). Titin mutations as the molecular basis for dilated cardiomyopathy. Biochemical and Biophysical Research Communications, 291(2), 385–393.
Matsumoto, Y., Hayashi, T., Inagaki, N., Takahashi, M., Hiroi, S., Nakamura, T., et al. (2005). Functional analysis of titin/connectin N2-B mutations found in cardiomyopathy. Journal of Muscle Research and Cell Motility, 26(6–8), 367–374. doi:10.1007/s10974-005-9018-5.
Moulik, M., Vatta, M., Witt, S. H., Arola, A. M., Murphy, R. T., McKenna, W. J., et al. (2009). ANKRD1, the gene encoding cardiac ankyrin repeat protein, is a novel dilated cardiomyopathy gene. Journal of the American College of Cardiology, 54(4), 325–333. doi:10.1016/j.jacc.2009.02.076.
Arimura, T., Bos, J. M., Sato, A., Kubo, T., Okamoto, H., Nishi, H., et al. (2009). Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 54(4), 334–342. doi:10.1016/j.jacc.2008.12.082.
Arimura, T., Hayashi, T., Matsumoto, Y., Shibata, H., Hiroi, S., Nakamura, T., et al. (2007). Structural analysis of four and half LIM protein-2 in dilated cardiomyopathy. Biochemical and Biophysical Research Communications, 357(1), 162–167. doi:10.1016/j.bbrc.2007.03.128.
Lange, S., Xiang, F., Yakovenko, A., Vihola, A., Hackman, P., Rostkova, E., et al. (2005). The kinase domain of titin controls muscle gene expression and protein turnover. Science, 308(5728), 1599–1603. doi:10.1126/science.1110463.
Puchner, E. M., Alexandrovich, A., Kho, A. L., Hensen, U., Schafer, L. V., Brandmeier, B., et al. (2008). Mechanoenzymatics of titin kinase. Proceedings of the National Academy of Sciences of the United States of America, 105(36), 13385–13390. doi:10.1073/pnas.0805034105.
Witt, C. C., Witt, S. H., Lerche, S., Labeit, D., Back, W., & Labeit, S. (2008). Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. The EMBO Journal, 27(2), 350–360. doi:10.1038/sj.emboj.7601952.
Zou, P., Pinotsis, N., Lange, S., Song, Y. H., Popov, A., Mavridis, I., et al. (2006). Palindromic assembly of the giant muscle protein titin in the sarcomeric Z-disk. Nature, 439(7073), 229–233.
Bos, J. M., & Ackerman, M. J. (2010). Z-disc genes in hypertrophic cardiomyopathy: stretching the cardiomyopathies? Journal of the American College of Cardiology, 55(11), 1136–1138. doi:10.1016/j.jacc.2009.12.016.
Chiu, C., Bagnall, R. D., Ingles, J., Yeates, L., Kennerson, M., Donald, J. A., et al. (2010). Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. Journal of the American College of Cardiology, 55(11), 1127–1135. doi:10.1016/j.jacc.2009.11.016.
Gunkel, S., Heineke, J., Hilfiker-Kleiner, D., & Knoll, R. (2009). MLP: a stress sensor goes nuclear. Journal of Molecular and Cellular Cardiology, 47(4), 423–425. doi:10.1016/j.yjmcc.2009.07.012.
Boateng, S. Y., Senyo, S. E., Qi, L., Goldspink, P. H., & Russell, B. (2009). Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. Journal of Molecular and Cellular Cardiology, 47(4), 426–435. doi:10.1016/j.yjmcc.2009.04.006.
Hayashi, T., Arimura, T., Itoh-Satoh, M., Ueda, K., Hohda, S., Inagaki, N., et al. (2004). Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. Journal of the American College of Cardiology, 44(11), 2192–2201. doi:10.1016/j.jacc.2004.08.058.
Knöll, R., Hoshijima, M., Hoffman, H. M., Person, V., Lorenzen-Schmidt, I., Bang, M. L., et al. (2002). The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell, 111(7), 943–955.
Knöll, R., Kostin, S., Klede, S., Savvatis, K., Klinge, L., Stehle, I., et al. (2010). A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circulation Research, 106(4), 695–704. doi:10.1161/CIRCRESAHA.109.206243.
Furukawa, T., Ono, Y., Tsuchiya, H., Katayama, Y., Bang, M., Labeit, D., et al. (2001). Specific interaction of the potassium channel beta-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. Journal of Molecular Biology, 313(4), 775–784.
Knöll, R., Postel, R., Wang, J., Kratzner, R., Hennecke, G., Vacaru, A. M., et al. (2007). Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation, 116(5), 515–525.
Postel, R., Vakeel, P., Topczewski, J., Knoll, R., & Bakkers, J. (2008). Zebrafish integrin-linked kinase is required in skeletal muscles for strengthening the integrin-ECM adhesion complex. Developmental Biology, 318(1), 92–101.
Herrmann, H., Bar, H., Kreplak, L., Strelkov, S. V., & Aebi, U. (2007). Intermediate filaments: from cell architecture to nanomechanics. Nature Reviews. Molecular Cell Biology, 8(7), 562–573. doi:10.1038/nrm2197.
Goldfarb, L. G., Olive, M., Vicart, P., & Goebel, H. H. (2008). Intermediate filament diseases: desminopathy. Advances in Experimental Medicine and Biology, 642, 131–164.
Klauke, B., Kossmann, S., Gaertner, A., Brand, K., Stork, I., Brodehl, A., et al. (2010). De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Human Molecular Genetics, 19(23), 4595–4607. doi:10.1093/hmg/ddq387.
Callaghan SC, White E (2007) The role of sarcomere and cytoskeleton in cardiac mechanostransduction. In: Weckstrom M, Tavi P (eds.), Cardiac Mechanotransduction. Springer Verlag, pp. 28–47.
Bloom, S., Lockard, V. G., & Bloom, M. (1996). Intermediate filament-mediated stretch-induced changes in chromatin: a hypothesis for growth initiation in cardiac myocytes. Journal of Molecular and Cellular Cardiology, 28(10), 2123–2127. doi:10.1006/jmcc.1996.0204.
Sullivan, T., Escalante-Alcalde, D., Bhatt, H., Anver, M., Bhat, N., Nagashima, K., et al. (1999). Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. The Journal of Cell Biology, 147(5), 913–920.
Chandar, S., Yeo, L. S., Leimena, C., Tan, J. C., Xiao, X. H., Nikolova-Krstevski, V., et al. (2010). Effects of mechanical stress and carvedilol in lamin A/C-deficient dilated cardiomyopathy. Circulation Research, 106(3), 573–582. doi:10.1161/CIRCRESAHA.109.204388.
Nikolova-Krstevski, V., Leimena, C., Xiao, X. H., Kesteven, S., Tan, J. C., Yeo, L. S., et al. (2010). Nesprin-1 and actin contribute to nuclear and cytoskeletal defects in lamin A/C-deficient cardiomyopathy. J Mol Cell Cardiol. doi:10.1016/j.yjmcc.2010.12.001.
Sadoshima, J., Xu, Y., Slayter, H. S., & Izumo, S. (1993). Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell, 75(5), 977–984.
Yasuda, N., Akazawa, H., Qin, Y., Zou, Y., & Komuro, I. (2008). A novel mechanism of mechanical stress-induced angiotensin II type 1-receptor activation without the involvement of angiotensin II. Naunyn Schmiedebergs Arch Pharmacol, 377(4–6), 393–399. doi:10.1007/s00210-007-0215-1.
Yasuda, N., Miura, S., Akazawa, H., Tanaka, T., Qin, Y., Kiya, Y., et al. (2008). Conformational switch of angiotensin II type 1 receptor underlying mechanical stress-induced activation. EMBO Reports, 9(2), 179–186. doi:10.1038/sj.embor.7401157.
Frank, D., Kuhn, C., Brors, B., Hanselmann, C., Ludde, M., Katus, H. A., et al. (2008). Gene expression pattern in biomechanically stretched cardiomyocytes: evidence for a stretch-specific gene program. Hypertension, 51(2), 309–318. doi:10.1161/HYPERTENSIONAHA.107.098046.
Acknowledgement
Dr. R. Knöll is supported by the DFG (Kn 448/9-1, Kn448/10-1), the Fritz Thyssen Foundation as well as by funds of the British Heart Foundation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Byambajav Buyandelger and Keat-Eng Ng are co-first authors.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Buyandelger, B., Ng, KE., Miocic, S. et al. Genetics of Mechanosensation in the Heart. J. of Cardiovasc. Trans. Res. 4, 238–244 (2011). https://doi.org/10.1007/s12265-011-9262-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-011-9262-6