
Abstract
Human’s robust cognitive abilities, including creativity and language, are made possible, at least in large part, by evolutionary changes made to the cerebral cortex. This paper reviews the biology and evolution of mammalian cortical radial glial cells (primary neural stem cells) and introduces the concept that a genetically step wise process, based on a core molecular pathway already in use, is the evolutionary process that has molded cortical neurogenesis. The core mechanism, which has been identified in our recent studies, is the extracellular signal-regulated kinase (ERK)-bone morphogenic protein 7 (BMP7)-GLI3 repressor form (GLI3R)-sonic hedgehog (SHH) positive feedback loop. Additionally, I propose that the molecular basis for cortical evolutionary dwarfism, exemplified by the lissencephalic mouse which originated from a larger gyrencephalic ancestor, is an increase in SHH signaling in radial glia, that antagonizes ERK-BMP7 signaling. Finally, I propose that: (1) SHH signaling is not a key regulator of primate cortical expansion and folding; (2) human cortical radial glial cells do not generate neocortical interneurons; (3) human-specific genes may not be essential for most cortical expansion. I hope this review assists colleagues in the field, guiding research to address gaps in our understanding of cortical development and evolution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
What makes us human? The answer lies in our cerebral cortex, as proposed by Pasko Rakic in 2006: “Within the past 125 years, we have witnessed great strides in understanding the development and evolution of the cerebral cortex, arguably the structure that makes us human” [1]. The cerebral cortex is the biological substrate of our unique cognitive abilities, including memory, complex reasoning, and advanced language.
The embryonic development of the vertebrate forebrain is guided by signals from three major patterning centers: a rostral patterning center that secretes fibroblast growth factors (FGFs), a caudodorsal center that secretes Wingless/Int proteins (WNTs) and bone morphogenetic proteins (BMPs), and a ventral center that secretes sonic hedgehog (SHH) [2,3,4,5,6,7,8,9,10,11,12,13,14]. These distinct signaling centers instruct the regional development of the telencephalon along the dorsoventral and rostrocaudal axes. Notably, recent studies have revealed that the topology of patterning signaling centers in the developing human telencephalon is similar to that in the mouse and ferret [15,16,17,18,19].
The cerebral cortex is derived from the dorsal telencephalon, or pallium, which includes the ventral, lateral, dorsal, and medial pallium [20, 21]. The WNT signaling pathway plays a major role in establishing the neurogenic program in the pallium [22,23,24,25]. It is believed that BMP and WNT signaling work together in early pallial (hippocampal and cortical) neuroepithelial cells to regulate their quiescence-proliferation balance [26,27,28,29,30]. WNT signaling is gradually downregulated in cortical radial glial (RG) cells after neurogenesis occurs, but WNT response transcription factor genes Pax6, Dmarta2, Emx2, and Lhx2 in the cortical RG cells continue to protect and promote the cortical neurogenic program [22, 31,32,33,34]. Cortical patterning and neurogenesis are also protected by the GLI3 repressor form (GLI3R)-mediated suppression from the ventralizing effects of SHH and FGF signaling [35,36,37,38,39,40,41,42].
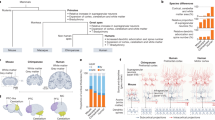
A distinct feature of mammals is their six-layered neocortex (new cortex), which is a specific feature of all mammals. In humans, ~90% of the cerebral cortex is neocortex. Over the course of evolution, the human cerebral cortex attained the largest number of neurons among primates (16.3 billion), which is thought to be the dominant contributor to underlying human intelligence. In contrast, the chimpanzee cerebral cortex has 8 billion neurons, the macaque cortex (old world monkey) has about 1.7 billion, the marmoset (new world monkey) cortex has 0.245 billion, the ferret cortex has 0.04 billion, and the mouse cortex has only 0.014 billion neurons [43,44,45,46] (Fig.1). Therefore, to be human, an essential step is to generate 16.3 billion cortical neurons.

Mammalian cortical expansion and evolution. Mammalian cortical neuron numbers range from 0.014 billion in mice to 16.3 billion in humans. Brain volumes range from 0.4 cc in mice to 1250 cc in humans. I suggest that there is a fundamental conserved genetic/biochemical engine that drives the increase in the number of cortical neurons, which further drives cortical size and brain volume expansion during evolution. Note that the mouse is lissencephalic but originated from a larger and gyrencephalic ancestor, as gyrencephalic ferrets separated before mice from the human phylogenetic tree.
Mouse and Human Cortical Radial Glial Cell Lineage Progression
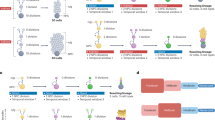
Mammalian cortical RG cells, known as cortical primary neural stem cells (NSCs), are the source of all cortical glutamatergic pyramidal neurons (PyNs), astrocytes, and the vast majority of oligodendrocytes [47,48,49,50,51,52,53] (Fig. 2a). During cortical development, cortical NSCs in mice primarily have one type of RG cell, known as full-span radial glial (fRG) cells [54, 55]. However, during human corticogenesis, there are three types of cortical RG cells: ventricular zone (VZ) fRG cells, which give rise to VZ truncated radial glia (tRG) and outer radial glia (oRG, also known as basal RG) cells, located in the outer subventricular zone (outer SVZ, OSVZ) [55,56,57,58,59,60,61,62,63,64,65,66]. Prior to cortical neurogenesis, cortical neuroepithelial cells divide symmetrically to amplify the founder pool. Once neurogenesis starts, these cells are believed to directly transform into cortical fRG cells, which then divide asymmetrically to self-renew and produce intermediate progenitor cells (IPCs) for PyNs (PyN-IPCs). Neurogenic fRG cells in the cortical VZ sequentially generate distinct subtypes of PyNs in an inside-out pattern, with deep-layer PyNs born first, followed by PyNs of upper layers (Fig. 2a, b) [50,51,52, 67,68,69].

Mouse and human cortical RG cell lineage progression. a, b Mouse cortical neurogenesis and gliogenesis. Upon entry into the neurogenic phase, a single mouse cortical fRG cell may divide 5-6 times and generate PyN-IPCs, which produce 8–9 PyNs distributed in both deep and upper layers. Cortical gliogenesis occurs after neurogenesis. At the gliogenic stage, mouse fRG cells first generate bMIPCs, which then give rise to cortical APCs, OPCs, and OBIN-IPCs. c Mammalian cortical PyNs are first generated in the ventral and lateral pallium, followed by the dorsal and medial pallium (cortex). Tbr1 and Neurod6 mRNA in situ hybridization on mouse brain sections at E12.5. Images are reprinted with permission from Moreau et al., 2021 [92]. d Human cortical neurogenesis and gliogenesis. I propose that a single human cortical neurogenic fRG cell may divide ~30 times and generate PyN-IPCs, and these PyN-IPCs may produce 30-45 PyNs distributed in deep layers. Around GW16, fRG cells give rise to tRG and oRG cells. A single human oRG cell in the lateral OSVZ may divide ~30 times and generate PyN-IPCs, and these PyN-IPCs may produce 30-45 PyNs distributed in upper layers. Human tRG cells undergo a neurogenesis-to-gliogenesis switch and generate bMIPCs, which divide several times and give rise to cortical APCs, OPCs, and OBIN-IPCs. e A single human cortical fRG cell may generate 60-90 PyNs. f, g Signaling pathways and transcription factors regulate mouse and human cortical RG cell neurogenesis, quiescence, proliferation, and self-renewal. Those signals that are stronger in human cortical RG cells, than in mice, are highlighted in magenta.
Cell lineage analysis shows that at the end of cortical neurogenesis, mouse cortical fRG cells progress from generating PyNs to astrocytes, oligodendrocytes, and a subpopulation of GABAergic olfactory bulb interneurons (OBINs) [48,49,50,51, 70,71,72,73,74,75,76,77,78,79]. However, there has been a fundamental gap in understanding how these diverse cell subtypes are generated. Our recent studies using genetic labeling methods in mice have demonstrated that during the cortical gliogenesis phase, fRG cells continue to divide asymmetrically to self-renew and produce a different type of IPC that is multipotent (tri-potential), which we have termed basal multipotent IPCs (bMIPCs) [80]. These bMIPCs undergo several rounds of mitosis and generate cortical astrocyte lineage-restricted IPCs (APCs), oligodendrocyte lineage-restricted IPCs (OPCs), and cortex-derived OBIN lineage-restricted IPCs (OBIN-IPCs) (Fig. 2a). These lineage-restricted IPCs then divide symmetrically to generate cortical astrocytes, oligodendrocytes and OBINs, respectively [80, 81].
Further research has shown that the molecular features of this process are consistent with known transcriptional mechanisms for generating oligodendrocytes, astrocytes, and OBINs. Cortical fRG cells generate bMIPCs in the SVZ that express epidermal growth factor receptor (EGFR), ASCL1, and OLIG2/1. When bMIPCs give rise to cortical APCs, they maintain the expression of EGFR and OLIG2/1 but downregulate ASCL1 expression, whereas OPCs maintain ASCL1 and OLIG2/1 expression but downregulate EGFR. Conversely, OBIN-IPCs maintain ASCL1 and EGFR expression but downregulate OLIG2/1 expression (Fig. 2a) [80,81,82].
In 1983, in vitro, studies demonstrated the existence of bi-potent glial-IPCs (O-2A cells) that give rise to both oligodendrocytes and astrocytes [83]. This study was not confirmed in vivo for a long time [84], but it is now accepted that both cortical OPCs and APCs are derived from the same population of IPCs, identified by us and others in the mouse and human cortex [60, 80, 85,86,87,88,89,90,91]. Furthermore, our genetic labeling experiments show that cortex-derived OBINs are also generated from these IPCs (bMIPCs) during development (Fig. 2a) [80, 85].
Cortical neurogenesis and gliogenesis always first occur in the rostroventral cortex, followed by the lateral, dorsal, and caudomedial cortex (Fig. 2c) [80, 92]. This is regulated by gradients of FGF, WNT-BMP, SHH signaling, and their contribution to the gradient expression of cortical patterning transcription factors (Nr2f1, Pax6, Sp8, Lhx2, and Emx2) in the cerebral cortex [4, 5, 22, 31,32,33,34, 93,94,95,96,97]. For example, neurogenesis occurs in the mouse ventrolateral cortex from E10.5 to E16.5, followed by gliogenesis, whereas neurogenesis in the medial cortex may occur from E12.5 to E18.5, after which gliogenesis begins.
In the human cerebral cortex, neuroepithelial cells start to convert into fRG cells around gestational week 7 (GW7) [59, 98, 99]. Then, fRG cells undergo asymmetric cell division to self-renew and produce PyN-IPCs around GW8, which may exclusively differentiate into deep cortical layer PyNs [56, 59, 60, 98, 100]. After approximately 9 weeks of neurogenesis (around GW16), human cortical fRG cells give rise to oRG and tRG cells [59, 60, 62]. oRG cells inherit the long basal fibers of fRG cells, while tRG cells inherit the apical domains of fRG cells [59, 60, 62, 101]. Both oRG and tRG cells can self-renew. The emergence of the cortical OSVZ in higher-order mammals is fundamentally different from that in mice [64]. oRG cells in the OSVZ mainly generate upper layer cortical PyNs [56, 57, 59, 61, 98, 102,103,104], and this process in humans lasts around 10 weeks from GW17 to GW26 (Fig. 2d, e) [55, 60].
In contrast to neurogenic oRG cells, our studies suggest that tRG cells in the cortical VZ primarily generate cortical glia [54, 55, 60]. After tRG cells are generated from fRG cells, a subpopulation of putative primed/active tRG cells starts to express EGFR and ASCL1. Initially, this population of primed tRG cells generates PyN-IPCs, but soon they undergo a neurogenesis-to-gliogenesis switch and generate bMIPCs that express ASCL1, EGFR, and OLIG1/2 [55, 60]. These bMIPCs then give rise to cortical APCs, OPCs, and OBIN-IPCs (Fig. 2d) [60]. Thus, this process in the human cortex is similar to that in the mouse cortex [60, 80], suggesting that mammalian cortical gliogenesis is evolutionarily conserved and that the presence of EGFR-expressing progenitors in the cortical VZ/SVZ is a strong signal for the onset of cortical gliogenesis in both mice and humans [55, 60, 80, 87].
As mouse cortex development proceeds after birth, fRG cells gradually give rise to human tRG-like NSCs, also known as B1 NSCs, which mainly generate OBIN-IPCs [70, 76, 105,106,107,108]. During aging, the number of NSCs and neurogenesis in the SVZ of the lateral ventricle declines [109], but the process of OBIN genesis persists throughout life in mice [106, 109]. On the other hand, while human postnatal cortical tRG cells (B1 NSCs) also generate OBIN-IPCs, this process only occurs during the first year of life. In the adult human SVZ, very few newly born OBINs are detected [110,111,112,113,114,115,116,117,118].
I propose that in both mouse and human brains, PyN genesis and gliogenesis first occur in the rostroventrolateral cortex, followed by the dorsal and caudomedial cortex [60, 80, 92]; this is a consistent rule in mammalian cortical development. For example, fRG cells in the human ventral cortex might start to generate PyNs at GW8, whereas fRG cells in the medial cortex might continue to expand their population by symmetric division and might not produce PyNs until GW10. At GW16, fRG cells in the ventral cortex give rise to oRG and tRG cells, whereas fRG cells in the medial cortex start to do so around GW18 [60]. Indeed, at GW18, we observe many human cortical APCs (GFAP+ HOPX+ cells) in the ventral cortex, whereas very few APCs are seen in the dorsal and medial cortex [60]. It is believed that the length of the human cortical neurogenic period is more than 130 days (GW8–GW26); however, a single fRG cell and its progeny oRG cell may generate cortical PyNs for only 110 days [55, 58, 98, 119, 120].
“Slow and Multistep” Processes Fundamentally Underlie Cortical Neurogenesis during Evolution
Asymmetric division of stem cells is the key mechanism to maintain the balance between self-renewal and differentiation. During the cortical neurogenic stage, RG cells undergo asymmetric divisions into one daughter cell equivalent to the RG cell itself and another daughter cell committed to becoming a PyN-IPC. In the developing mouse cortex, a single neurogenic fRG cell divides 5-6 times and produces 8–9 PyNs that are distributed in both deep and upper layers (Fig. 2a, b) [48]. Mouse cortical neurogenic fRG cell cycle length ranges from 18–26 hours [120,121,122,123]. Compared with other mammals, mouse cortical fRG cells have a short cell cycle and short neurogenic period. Mice may also have the shortest length of gestation among mammals [120]. As noted above, individual mouse cortical fRG cells only have 6 days to generate PyN-IPCs and then switch to cortical gliogenesis (Fig. 2a) [48, 49, 80, 85, 91], resulting in mouse cortical fRG cells not undergoing strict self-renewal because their potential progressively changes and they generate different progenies over the course of cortical neurogenesis (Fig. 2a, b) [48,49,50,51, 75, 124].
In contrast to mice, non-human primate and human cortical RG cell cycles can be up to 44-63 hours, and the lengthening of cortical RG cell cycles is a well-documented feature of primates with larger brains [66, 120, 125,126,127]. Human cortical RG cells likely have the longest cell cycle and the longest neurogenic period. Humans also have the longest gestation period among primates [119, 120, 128]. Longer cell cycles are believed to maintain genomic fidelity, reduce the rate of somatic mutation, regulate cell fate, and ensure stem cell lineage fidelity [129, 130]. Therefore, human cortical RG cells undergo much stricter self-renewal than mice (Fig. 2d, e). I propose that the lengthening of the cortical RG cell cycle is an essential prerequisite for increasing the RG neurogenic period, whereas shortening the RG cell cycle results in shortening the neurogenic period and initiating cortical gliogenesis. Longer cell cycles and longer neurogenic periods of cortical RG cells inevitably promote a “slow and multistep” manner of neurogenesis in the human cortex. For example, a single neurogenic human cortical fRG cell in the lateral cortex may divide approximately 30 times (from GW8 to GW16) and generate PyN-IPCs, which may produce 30-45 PyNs distributed in deep layers. Around GW16, fRG cells give rise to tRG and oRG cells. A single human oRG cell in the lateral OSVZ may also divide 30 times (from GW16 to GW24) and generate PyN-IPCs, which may produce 30-45 PyNs distributed in upper layers (Fig. 2d, e). In the mouse cortex, a single cortical fRG cell may produce only 2 PyNs in layer 6, whereas a single human cortical fRG cell may produce more than 20 PyNs in layer 6, which exhibit similar morphological, physiological, molecular, and connectional features (Fig. 2d, e), and then they start to generate PyNs in layer 5. The acquisition of a “slow and multistep” manner of cortical neurogenesis is therefore a fundamental property that leads to the evolution of the human cortex.
What are the molecular mechanisms underlying the lengthening of cell cycles and the neurogenic period of human cortical RG cells? We and others have looked for fundamental molecular mechanisms that regulate both mouse and human cortical development and neurogenesis (Fig. 2f, g). For example, cortical neurogenesis is protected by GLI3R-mediated suppression from the ventralizing effects of SHH and FGF signaling [35,36,37,38,39,40,41,42]. Pax6, Dmarta2, Emx2, and Lhx2 expression in the cortical RG cells promote and protect neurogenesis [22, 31,32,33,34, 131]. NOTCH signaling regulates cortical RG cell quiescence and self-renewal [56, 132,133,134,135]. FGF-ERK signaling promotes cortical RG cell proliferation and self-renewal [12, 54, 93, 136,137,138,139,140,141]. SHH signaling, especially in mice, may promote cortical neurogenic RG cell proliferation [55, 142,143,144] (Fig. 2f).
While the transcriptional profiles of human and mouse neocortical RG cells are broadly conserved during neurogenesis, studies have also identified some signaling pathways that are much stronger in human cortical RG cells. Among these signaling pathways, FGF-ERK, BMP7-SMAD, LIF/LIFR, GLI3R, and PDGFD–PDGFRB signaling are stronger in humans than in mice (Fig. 2g) [54, 55, 58, 145,146,147]. FGF-ERK signaling is required for normal RG cell proliferation and self-renewal. On the other hand, BMP7-SMAD, LIF/LIFR, GLI3R, and PDGFD–PDGFRB [148] signaling pathways play crucial roles in maintaining human cortical RG cell self-renewal and quiescence. Moreover, BMP7-SMAD signaling inhibits cortical gliogenesis, which may greatly increase the human cortical neurogenic period [54, 55].
As mentioned above, oRG cells in the OSVZ mainly generate upper cortical layer PyNs [56, 57, 59, 61, 98, 102,103,104], and this process in humans lasts around 7-10 weeks from GW16 to GW26 (Fig. 2d–g) [55, 60]. This conclusion has been supported and expanded upon by the analysis of the transcriptome of human oRG and tRG cells using single-cell RNA sequencing (scRNA-Seq) (Fig. 3) [55, 58]. To define the major transcriptional features of gliogenic tRG and neurogenic oRG cells, we performed differential gene expression analysis between neurogenic oRG cells and gliogenic tRG cells in GW22, GW23, and GW26 cortex [88]. Although both human cortical oRG and tRG cells are derived from fRG cells, they exhibit distinct transcriptional signatures. tRG cells have higher CXCL12/CXCR4, SHH, and EGFR signaling, which promotes cortical gliogenesis (Fig. 3a, e, f) [85, 149,150,151,152,153]. On the other hand, higher BMP7, GLI3R, CXCL14/CXCR4, and FGF-ERK signaling in oRG cells promote self-renewal and quiescence and protect neurogenesis (Fig. 3e) [28, 54, 55, 142]. Notably, strong expression of BMP7-SMAD signaling, and its downstream target CXCL14, are absolutely required for maintaining stem cell identity [154, 155]. CXCL14/CXCR4 signaling may also block CXCL12/CXCR4 signaling [156].

Human cortical oRG cells and tRG cells exhibit distinct transcriptional signatures. a Schematic summarizing the process of mouse and human cortical RG neurogenesis and gliogenesis. b Immunohistochemical analysis shows that human oRG cells have a larger soma (more than 10 µm in diameter, arrowheads) and do not express EGFR. Note that HOPX+ EGFR+ cells are cortical APCs, as they have smaller soma size. c PAX6+ oRG cells produce TBR2 (EOMES)+ PyN IPCs that maintain PAX6 expression (arrows). d Very strong HOPX-expressing oRG cells generate TBR2 (EOMES)+ PyN IPCs (arrowheads) that never express HOPX, suggesting that oRG cells have longer cell cycles. e scRNA-Seq analysis of molecular profiles of human cortical RG cells at GW22, GW23, and GW26. Heat map of selected differentially expressed genes for oRG cells versus tRG cells. f Selected GO terms supporting that oRG cells are neurogenic while tRG cells are gliogenic. Images (a, e, f) are reprinted with permission from Li et al., (2024) [55]. Images (b–d) are adapted from Yang et al., 2022 [60].
Neurogenesis in human cortical oRG cells is further slowed down. Our immunohistochemical analyses found that a subpopulation of human cortical tRG cells express EGFR, whereas oRG cells do not (Fig. 3b) [60]. scRNA-Seq analysis of fluorescence-activated cell sorting EGFR+ cells isolated from the frontal lobe of the human cerebral cortex between GW21 and GW26 supported this conclusion [60, 87]. We also observed that strong HOPX-expressing oRG cells with large soma size (more than 10 µm in diameter) generate PyN-IPCs that express EOMES (TBR2) but never express HOPX (Fig. 3c, d) [60], further suggesting that oRG cells have a longer cell cycle and undergo stricter self-renewal. EOMES-expressing PyN-IPCs also express PPP1R17, which regulates G1/S transition and slows PyN-IPC cell cycle progression [119]. All these observations reveal that oRG neurogenesis follows a typical “slow and multistep” manner.
In vitro studies have shown that while enhancing BMP signaling alone promotes terminal astrocytic differentiation, exposure to both BMP and FGF2 (ERK signaling) maintains the stem cell character of cultured progenitor cells [157, 158]. We speculate that higher FGF-ERK activity in oRGs antagonizes quiescent BMP7 signaling through phosphorylation of the SMAD linker domain [159, 160]. oRG cells also express higher levels of NOG, a secreted BMP antagonist [55, 58]. Therefore, the intricate balance of quiescence, proliferation, self-renewal, and differentiation (neurogenesis) is well controlled by those signaling pathways that are at higher levels in human cortical oRG cells, resulting in oRG cells playing a key role in the increased generation of PyNs that constitutes the foundation of neocortex expansion. At the end of cortical neurogenesis, oRG cells may directly transform into astrocytes, but not generate OPCs [60], because BMP signaling is further enhanced in the cortex from the second trimester to the third trimester [55].
ERK Signaling Drives the Evolutionary Expansion of the Mammalian Cerebral Cortex
The human cerebral cortex has the largest number of neurons (16.3 billion) among primates (Fig. 1). Two main factors determine cortical PyN numbers and the surface area of the cerebral cortex. One is the size of the cortical neuroepithelial and fRG cell pool present in the VZ at the beginning of cortical neurogenesis, which largely determines the PyN number. This is known as “the radial unit hypothesis” originally proposed by Rakic [1, 161,162,163]. The second is the length of the cortical neurogenic period [120, 126, 164]. As indicated above, humans have the longest cortical neurogenic period [55, 59, 60, 119, 120], allowing cortical RG cells to produce more PyNs. According to the “radial unit hypothesis” model, an increase in the neurogenic period appears to contribute a linear increase in the number of cortical PyNs within each radial cortical column (Fig. 2b, e), which also contributes to an increase in cortical thickness [165,166,167,168].
Remarkable progress has been made over the past thirty years in identifying potential mechanisms that contribute to mammalian cortical development, expansion, and evolution [1, 14, 46, 50,51,52, 68, 98, 102, 120, 126, 146, 161, 163, 169,170,171,172,173,174,175]. However, unifying molecular principles, if they exist, have not yet been identified that have driven progressively larger cortices during mammalian evolution. As indicated in Fig. 1, I believe that there must be a common rule that drives mammals to generate more and more cortical PyNs during evolution, which contributes to an expansion of the cortex and an increase in brain volume across primates [176,177,178]. I postulate, that we may have recently identified such a core mechanism that drives mammalian cortical expansion during evolution [54].
By comparing mouse, ferret, monkey, and human developing cortex, we found that BMP7 is expressed by increasing numbers of cortical RG cells [55]. We demonstrated that BMP7-SMAD signaling represses EGFR expression in cortical RG cells and inhibits SHH signaling by promoting GLI3R formation [54, 55]. Therefore, BMP7 in cortical RG cells promotes neurogenesis, inhibits gliogenesis, and thereby extends the neurogenic period, resulting in the production of a large number of cortical PyNs. As mentioned above, BMP signaling also functions to maintain stem cells in a quiescent state and sustain stem cell self-renewal [28, 154, 155, 179,180,181,182].
What signals induce BMP7 expression in cortical RG cells during development and evolution? It turns out that FGF-ERK signaling is crucial. FGF signaling is required for neural induction, which begins during early gastrulation. The anterior neural ridge (ANR) is located in the anterior neural plate of vertebrates and expresses FGFs (FGF8/17/18), which are important for patterning the telencephalon [183,184,185]. This conserved mechanism dates back at least 500 million years, as the ANR-like genetic regulatory program of FGF signaling is present in hemichordates and amphioxus (Fig. 4a) [186]. The ANR-derived rostral telencephalic patterning center (RPC) produces FGFs (Fig. 4b, c) [7,8,9, 184, 187]. FGFs and their downstream targets, Spry1 and Spry2, are mainly expressed in the presumptive septum and their expression extends into the mouse medial cortex during early cortical neurogenesis [7, 141, 187]. Interestingly, Bmp7 is also expressed in the medial cortex [55, 188]. Moreover, previous studies have shown co-expression of Fgf8 and Bmp7 in the mouse ANR [9]. These observations indicate that Bmp7 expression might be induced by FGF signaling. Indeed, when we conditionally overexpressed Fgf8 in the cortex using hGFAP-Cre transgene lines, strong Bmp7 and HOPX expression was observed in the E14.5 neocortical VZ [54].

ERK signaling drives the evolutionary expansion of the mammalian cerebral cortex. a, b The ANR expressed FGFs in hemichordates (500 million years ago) and mice. c During mouse early forebrain development, FGFs are expressed in the rostral patterning center, whereas SHH is expressed in POA stem/progenitor cells and MGE-derived neurons. d, e At the beginning of cortical neurogenesis in the most recent ancestor to all mammals, it is assumed that there was already a subset of cortical fRG cells that expressed relatively higher levels of pERK. Elevated ERK signaling in these cortical fRG cells promotes BMP7 expression, which increases GLI3R generation and represses SHH signaling. A decrease in SHH signaling in cortical fRG cells further enhances ERK signaling. Therefore, the ERK-BMP7-GLI3R-SHH signaling pathway in cortical fRG cells participates in a positive feedback loop, which expands the cortical fRG cell pool, increases the length of the cortical neurogenic period, and thus can drive increases in cortical surface area and neurogenesis; this may underlie these features characteristically observed in evolutionarily more advanced mammals. f SHH signaling underlies cortical evolutionary dwarfism in mice. During mouse cortical development and evolution, smaller brains with smaller lateral ventricle volumes may result in higher SHH concentration in the cerebral spinal fluid and thus can increase SHH signaling in the cortex, which antagonizes ERK signaling. Relatively weak ERK signaling fails to induce Bmp7 expression in the dorsal cortical RG cells, resulting in a shortened period of cortical neurogenesis (for example, from more than 130 days in humans to about 7 days in mice). g ERK signaling is elevated in human cortical fRG and oRG cells during development and evolution and induces BMP7 expression that antagonizes SHH signaling. Images are reprinted with permission from Sun et al., 2024 [54].
The interaction of FGFs with FGFRs results in phosphorylation and nuclear translocation of ERK1/2, which phosphorylates target transcription factors [139, 189,190,191,192,193]. ERK also directly regulates gene expression via its DNA binding activity [194,195,196]. ERK signaling can be activated in the RosaMEK1DD mouse [151, 197, 198], which contains a Cre-inducible constitutively active rat Map2k1 allele. We introduced the hGFAP-Cre allele into the RosaMEK1DD mice and observed that strong activated ERK induced Bmp7 expression in the E14.5 cortical VZ (Fig. 5a–f). Furthermore, HOPX, ETV5, EGFR, and OLIG2 expression were also strongly upregulated (Fig. 5a-f). We have also shown that loss of cortical ERK signaling results in loss of Bmp7 and HOPX expression [54], premature neural differentiation, and depletion of cortical RG cells [54, 136,137,138]. Therefore, ERK signaling is required for cortical expression of Bmp7 and HOPX and maintaining self-renewal and the undifferentiated state of cortical RG cells. Notably, abnormal sustained FGF-ERK activity also induces a switch from cortical neurogenesis to gliogenesis [54, 140, 151, 199].

Constitutive ERK signaling induces Bmp7 expression in cortical RG cells and can drive cortical expansion. a–f Expression of pERK (phosphorylated ERK), BMP7, HOPX, ETV5, EGFR, and OLIG2 is increased in the cortex of hGFAP-Cre; RosaMEK1DD mice at E14.5 (arrows); reprinted with permission from Sun et al., 2024 [54]. g Expression patterns of Fgfr1, Fgfr2, and Fgfr3 in E12-E13 mouse cerebral cortex; reprinted with permission from Iwata and Hevner (2009) [190]. Note the low rostral-high caudal Fgfr3 expression gradient. h The mutational spectrum of human FGFR3. Gain of function FGFR3 mutations result in dwarfism syndromes, ranging from achondroplasia and hypochondroplasia to the more severe neonatal lethal thanatophoric dysplasias TDI and TDII; reprinted with permission from Ornitz and Legeai-Mallet (2015) [204]. i–k Temporal lobe enlargement and abnormal sulci in GW18, GW22, and GW40 TD fetuses; reprinted with permission from Hevner (2005) [205]. i, m Brain morphology of TD mouse at P0 and P28. Note that TD mice have an enlarged cerebral cortex; reprinted with permission from Lin et al., (2003) [211].
In the E11 mouse cortex, FGF-ERK signaling exhibits a rostral-high to caudal-low gradient, whereas SHH signaling exhibits a ventral-high to dorsal-low and caudal-high to rostral-low gradient, which is opposite (Fig. 4b–d) [2, 4, 5, 200]. Gain and loss of function studies demonstrated that SHH-SMO signaling antagonizes ERK-BMP7 signaling in mouse cortical fRG cells and sustained SHH-SMO activity also promotes the onset of cortical gliogenesis [54, 55, 85, 152, 201], suggesting that SHH signaling contributes to the shortening of the mouse cortical neurogenic period.
By integrating analyses of published mouse, ferret, monkey, and human cortical scRNA-Seq datasets with immunostaining and mRNA in situ hybridization experiments, we have provided strong evidence that ERK signaling is progressively higher in cortical RG cells in evolutionarily more advanced species [54]. We propose that similar to the mouse cortex, cortical RG cell expression of BMP7 in other mammals, including humans, is also induced by ERK signaling, consistent with BMP7 expression gradually upregulating in mouse, ferret, monkey, and human cortical RG cells during evolution [54, 55, 146, 147, 202]. Based on these findings, I believe that we have identified a core mechanism that drives cortical expansion and evolution.
We propose that ERK signaling drives the evolutionary expansion of the mammalian cerebral cortex (Fig. 4d–g) [54]. The most recent ancestor to all mammals, approximately 100 million years ago, is assumed to have already been somewhat gyrencephalic (Fig. 1) [120, 203], and is assumed to have had a subset of fRG cells in the developing dorsal cortex that expressed relatively higher levels of ERK. We showed that elevated ERK signaling in cortical fRG cells induces BMP7 expression, which then promotes GLI3R production and represses SHH signaling [54, 55]. A decrease in SHH signaling in cortical fRG cells further enhances ERK signaling. Therefore, ERK-BMP7-GLI3R-SHH signaling in cortical fRG cells participates in a positive feedback loop (Fig. 4d, e), which increases the size of the cortical RG cell pool, restrains but allows neurogenesis while inhibiting gliogenesis, and thus increases the length of the neurogenic period. We hypothesize that SHH concentration in cerebrospinal fluid is reduced in the enlarged lateral ventricle of big-brained mammals during cortical development and evolution; this could contribute to a further decrease in SHH signaling and an increase in ERK-BMP7-GLI3R signaling in cortical fRG cells (Fig. 4g).
Similarly, ERK-BMP7-GLI3R expression in oRG cells in the cortical OSVZ continues to participate in a positive feedback loop because the primary cilia of oRG cells do not contact cerebrospinal fluid in the lateral ventricle and thus may not receive SHH signaling. Indeed, scRNA-Seq analysis of the human cortex at GW18, 22, 23, and 26 [88], never reveals SHH target gene GLI1 expression in human oRG cells, whereas GLI1 expression is detected in tRG cells. SHH-SMO signaling promotes EGFR expression, whereas BMP7 and GLI3R repress EGFR expression in cortical RG cells [54, 55]. This is why tRG cells express EGFR, but oRG cells do not [55, 60, 87]. As mentioned above, EGFR-expressing primed/active tRG cells in the cortical VZ are mainly gliogenic, while oRG cells in the cortical OSVZ continue to generate PyNs. This two-germinal-zone system in the human developing cortex ensures that the onset of tRG gliogenesis occurs in the VZ, while oRGs in the OSVZ produce PyNs [54, 55, 60], the latter increases the length of the neurogenic period enabling the generation of more PyNs [120, 126, 164].
The lissencephalic mouse is believed to have originated from a larger and gyrencephalic ancestor (Fig. 1) [120, 203]. Based on our model, I propose that SHH signaling drives the phyletic dwarfism of the mouse cortex. Smaller brains with smaller lateral ventricles may result in higher concentrations of SHH that bathe the cortical VZ and antagonize ERK signaling during development and evolution (Fig. 4d, f), resulting in relatively lower ERK-BMP7 signaling in mouse cortical fRG cells [54], which reduces the size of the cortical RG cell pool and shortens the neurogenic period. I think that this is the major reason that the mouse cortex only has ~13.7 million neurons, whereas the human cortex has ~16.3 billion neurons (Fig. 1) [43,44,45].
Our proposed principle of cortical development, expansion, and evolution is supported by multiple types of genetic evidence. The normal development and expansion of the human cortex provide strong evidence that ERK signaling drives the evolutionary expansion of the mammalian cortex. The rostral patterning center releases FGFs, and promotes the size of the rostral cortex and basal ganglia [2, 4, 7, 8, 10,11,12, 14]. FGF-ERK-BMP7 signaling is elevated during cortical development and evolution, playing a crucial role in expanding the size of the RG cell pool and increasing the length of the neurogenic period [54, 55], resulting in a significant expansion of the human frontal cortex, the most highly developed and expanded brain structure in mammal evolution [1, 102].
Probably the strongest genetic evidence that supports our principle is the development and expansion of the human temporal and occipital cortex in Thanatophoric Dysplasia (TD) patients (Fig. 5g–k). TD is a lethal form of short-limbed dwarfism caused by mutations of FGFR3, which lead to constitutive activation of FGFR3 tyrosine kinase activity [190, 204, 205]. FGFR3 is expressed in a high caudomedial-low rostrolateral gradient in the human [206], ferret [207], and mouse cortical neuroepithelial and RG cells (Fig. 5g, h) [11, 208]. FGFR3 gain of function mutation in TD patients significantly strengthens ERK signaling [209, 210], which may further enhance BMP7 expression, causing the significant expansion of the temporal and occipital lobes in TD patients [190, 205]. Notably, the TD mouse provides an excellent model for the human TD, as they also exhibit the expansion of the cortex (Fig. 5l, m) [204, 209, 211,212,213,214,215], further providing evidence that FGF-ERK signaling can drive cortical expansion during evolution.
Finally, the case report that heterozygous loss of function mutations in the human BMP7 gene cause small head size, provides additional genetic evidence for BMP7’s function in promoting cortex size [216].
SHH Signaling is Not a Key Regulator of Primate Cortical Expansion and Folding
Based on evidence that SHH-SMO signaling antagonizes ERK-BMP7 signaling in mammalian cortical RG cells [54, 55], I propose that SHH signaling drives cortical evolutionary dwarfism, exemplified by the lissencephalic mouse that originated from a larger and gyrencephalic ancestor [120, 203]. However, several studies have concluded, contrary to my proposal, that SHH signaling promotes cortical expansion and gyration [217,218,219,220,221,222,223,224,225,226,227,228,229,230]. It is well known that SHH is a strong mitogen and promotes the proliferation of NSCs and IPCs in the brain [4, 85, 143, 152, 218, 231]. For example, SHH signaling promotes the extensive and prolonged symmetric division of granule cell precursors, which then differentiate into mature cerebellar granule cells in the cerebellum, the most numerous neurons of the brain [232]. Dysregulation of the SHH pathway results in SHH-subtype medulloblastoma, one of the most common malignant brain tumors in children [233]. In the wild-type mouse brain, the cortical VZ/SVZ expresses higher levels of Gli1 during the first postnatal week than before birth, indicating an increase in SHH signaling in the postnatal cortical VZ/SVZ [234]. This coincides with an increase in cortical OBIN genesis in the cortical SVZ after birth. Higher levels of SHH signaling are necessary for a Type C cell (derived from B1 NSCs) to produce 16-32 OBINs [235,236,237]. Therefore, SHH’s function does not appear to be part of the ERK-BMP7-GLI3R pathway that promotes the expansion of cortical PyN. Indeed, when SHH-SMO signaling is constitutively active in the cortex of hGFAP-Cre; RosaSmoM2 mice, although the proliferation of cortical fRG cells and the cortical VZ length are increased (Fig. 6a), a large number of EGFR+ OLIG2+ cells are also generated (these cells are not present in the cortex of control mice at E14.5) (Fig. 6a), indicative of a switch from cortical neurogenesis to gliogenesis. At E16.5, while more EOMES+ PyN-IPCs are present in the medial cortex, these cells are reduced in the lateral cortex as more EGFR+ OLIG2+ cells are generated in this region (Fig. 6b). At E17.5, cortical fRG cells precociously generate large numbers of OBIN-IPCs in the SVZ of hGFAP-Cre; RosaSmoM2 mice (Fig. 6c, d) [54, 55, 85]. We also ectopically overexpressed non-cholesterol-modified SHH (SHHN) in the E13.5 cortical VZ using in utero electroporation, and examined the cortex at E18.5. Similar to hGFAP-Cre; RosaSmoM2 mice, we observed a large number of cells expressing OBIN markers [85].

Constitutive SHH-SMO signaling promotes cortical OBIN genesis. a–d Higher levels of SHH-SMO signaling promote cortical RG cell and IPC proliferation, induce EGFR and OLIG2 expression, expand the cortical VZ/SVZ, and strongly induce GSX2 and DLX2 expression, favoring the generation of OBINs. Images are reprinted with permission from Sun et al., (2024) [54], Li et al., (2024) [55], and Zhang et al., (2020) [85].
After birth, in hGFAP-Cre; RosaSmoM2 mice, abnormal folding is observed in the medial cortex, the number of PyNs in the upper layer is increased, whereas the number of PyNs in the deep layer is reduced, and the thickness of the cortical SVZ is significantly expanded because OBIN-IPCs are robustly proliferating (Fig. 7a, b) [85, 218]. Therefore, higher levels of SHH signaling in the developing mouse cortex greatly disrupt the normal program of corticogenesis (Fig. 7a, b) [54, 55, 85, 201, 238]. This provides evidence against the idea that increasing SHH-signaling may underlie the evolutionary expansion of the primate cortex.

Constitutive SHH-SMO signaling promotes cortical abnormal folding. a Higher levels of SHH-SMO signaling reduce the number of TBR1+ PyNs in the deep cortical layers in hGFAP-Cre; RosaSmoM2 mice at P0. Note that the thickness of the SVZ is expanded and the cortex is enlarged. b Higher levels of SHH-SMO signaling induce abnormal medial cortical folding and cortical expansion, reduce the number of TBR1+ PyNs in the deep cortical layers, and increase SATB2+ PyNs in the upper cortical layers at P10; reprinted with permission from Wang et al., (2016) [218].
Finally, I have noted that abnormal cortical folding is observed in the medial cortex in transgenic mouse models used for studying cortical expansion, including hGFAP-Cre; RosaSmoM2 [218], Emx1-Cre; Gli3 F/F [239], Emx1-Cre; Cep83 F/F [240], Nes-Cre; Gpr161 F/F [226], and hGFAP-Cre; Pik3caH1047R (conditional activating mutations of Pik3ca) [241, 242] mouse lines. What are the molecular mechanisms underlying this folding phenotype? I suggest that because mouse medial cortical fRG cells express higher levels of ERK-BMP7-GLI3R signaling (similar to human cortical fRG and oRG cells) when they receive abnormal cell proliferation signals such as strong SHH signaling, they are able to sustain their self-renewal, maintain neurogenesis, and inhibit gliogenesis for a longer time than lateral cortical RG cells [54, 55], resulting in abnormal folding in the medial cortex.
Human Cortical RG Cells Do Not Generate Cortical Interneurons
The mammalian neocortex contains two main neuronal subpopulations: cortical PyNs and cortical inhibitory GABAergic interneurons (CINs). In the mouse cortex, PyNs constitute about 80%, whereas CINs constitute 20% of all cortical neurons. Recent studies in mouse models and humans provide evidence that CIN dysfunction is associated with neurological and psychiatric diseases such as epilepsy, schizophrenia, bipolar disorders, and autism, highlighting their crucial roles in the development and organization of cortical networks that underlie a wide range of cortical and mental functions [243,244,245,246,247,248,249,250,251]. Studies over the past 30 years have demonstrated that nearly all mouse CINs originate from subcortical ganglionic eminences (GEs)-the medial and caudal ganglionic eminence (MGE and CGE) and preoptic area (POA) [251,252,253,254,255,256,257,258,259,260,261,262,263,264,265,266]. The lateral ganglionic eminence (LGE) may not generate CINs, as the dorsal LGE mainly generates OBINs and the ventral LGE mainly generates striatal projection medium spiny neurons [131, 258, 264, 267,268,269,270,271,272,273,274,275,276,277,278,279,280,281].
Although the LGE, MGE, and CGE of the human fetal brain have similar morphological features as mice [282], the origin of CINs in humans and non-human primates was controversial as previous studies reported that, in contrast to rodents, a large number of CINs in humans and non-human primates are generated from cortical progenitors [283,284,285,286,287]. In 2013, two papers published back-to-back revealed that similar to mice, most, if not all, human CINs are also derived from the subcortical GEs [110, 288]. They showed that markers of the MGE (NKX2-1 and LHX6), LGE (SP8 in the dorsal LGE and ISL1 in the ventral LGE), and CGE (NR2F2 and SP8) were very similar in the developing human, macaque monkey and mouse telencephalon. Monkey and human fetal brain slice cultures and BrdU labeling provided strong evidence that primate CINs are derived from GEs [110, 288]. These results show that the patterns of transcription factor expression and the tangential migration of CINs from the GEs to the cortex are conserved from rodents to primates during mammalian brain evolution [110, 288,289,290].
In 2019, a group of scientists in the field published a review paper [102] and commented on mammalian CIN generation: “experiments in rodents have demonstrated that inhibitory interneurons are produced in the GEs and tangentially migrate into the cortex, and to a large extent, the human fetal brain appears to share fundamental mechanisms for CIN development [290]. Landmark publications [110, 288] challenged the earlier notion that up to 65% of CINs may derive from the dorsal pallium [283] and demonstrated a predominantly ventral pallial origin for interneurons”. In 2021, two studies [291, 292], by conducting scRNA-Seq and in situ sequencing in developing human brains, further revealed that transcriptional programs that control interneuron specification, migration, and differentiation are evolutionarily conserved in mice and humans.
More recently, several papers have claimed the discovery of a cortical origin of some human CINs [293,294,295,296,297,298]. However, these results are inconsistent, and some are even contradictory. For example, previous studies showed that when dissociated cells from GW15.5 cortical OSVZ and VZ/SVZ were cultured with BrdU to label proliferating cells, after 7 days, BrdU+ GABA+ cells were not observed [56], suggesting that cortical progenitors do not produce inhibitory neurons around GW15-GW16. In contrast, lentiviral clone labeling of dissociated cortical cells from GW15 to GW18 indicated that individual human cortical progenitors generate both PyNs and CINs, and these cortex-derived interneurons were transcriptionally similar to CGE-derived CINs [293]. Another study using cell-type-resolved mosaicism revealed that a substantial portion of cortex-derived interneurons may contribute to parvalbumin+ CINs (MGE-like), whereas cortical NR2F2 (COUP-TFII)+ interneurons are most likely derived ventrally from the CGE [298].
In addition, some papers reported that human cortical neurogenesis ceases around GW19 [299], and reported that cortical oRG cells give rise to CGE-like CINs and/or cortical OPCs [86, 89, 295,296,297, 300]. Again, these results do not make sense as cortical oRG cells are mainly neurogenic and do not receive SHH signaling. In the mouse cortex, when SHH-SMO signaling is absent (hGFAP-Cre; SmoF/F), the presence of EGFR+ ASCL1+ OLIG2+ bMIPCs from cortical fRG cells is postponed, cortical gliogenesis is retarded, the cortical PyN genesis period is prolonged [54, 55], and cortical fRG cells fail to generate OBINs [85]. This is consistent with previous observations that SHH signaling is required for the generation of interneurons and oligodendrocytes in the ventral GEs [231, 301, 302]. In the mouse dorsal cortex, SHH signaling appears to favor the specification of OBINs but not CINs [85, 234, 303].
To further evaluate the origins of human CINs, I have re-examined mouse and human cortical scRNA-Seq data and found that nearly all CINs are derived from the subcortical GEs (Fig. 8, 9). Here is the basic logic of this analysis framework. As mentioned above, in mice, CINs appear to be exclusively derived from the subcortical GEs. In humans, if a subset of CINs is derived from cortical RG cells, we should be able to identify IPCs for these CINs in the developing human cortex, and these IPCs must express DLX1 and DLX2 [304, 305]. It is worth noting that a number of IPCs in the human and mouse subcortical GEs express EGFR, but when EGFR-expressing IPCs and their progeny migrate into the cortex, they downregulate EGFR expression [60, 80]. Therefore, all EGFR+ cells seen in the mouse and human cortex are produced directly or indirectly from cortical RG cells. I re-analyzed scRNA-seq datasets from a total of 98,047 mouse cortical cells obtained from mouse E10.5 to P4 cortex [306], and a total of 57,868 human cortical cells obtained from human GW18 (19,373 cells), GW22 (12,567 cells), GW23 (12,557 cells), and GW26 (13,371 cells) cortical tissue [88]. I found that, first, it is easy to identify MGE-, POA-, and CGE-derived DLX1+, and/or DLX2+, GAD2+, DLX5+, SP8+, SP9+, NR2F2+, LHX6+ CIN clusters in both mice and humans (Fig. 8a, 9a). Second, only a small number of IPCs for interneurons are identified in the mouse and human cortical cells, and they express EGFR, GSX2, DLX1, DLX2, and GAD2 (Fig. 8a, 9a). These IPCs are adjacent to the cortical bMIPC cluster that express ASCL1, EGFR, OLIG1, and OLIG2, demonstrating that these GSX2+, DLX1+, DLX2+, GAD2+, and EGFR+ IPCs are likely to be OBIN-IPCs, but not CIN-IPCs. scRNA-Seq analysis of fluorescence-activated cell sorting EGFR+ cells isolated from the frontal lobe of the human cerebral cortex between GW21 and GW26 supported these observations [60, 87]. Immunostaining further validated EGFR+ GSX2+ cells and EGFR+ DLX2+ cells in the GW23 cortex [60]. Notably, GSX2+ cells in the mouse cortex are bMIPCs that are able to produce cortical APCs, OPCs, and OBIN-IPCs in vivo [85], but favor the specification of OBIN identity [307, 308]. Therefore, human cortical tRG cells and mouse cortical fRG cells indeed produce inhibitory interneurons, but these interneurons are for the OB, not for the cortex (Fig. 8b–d, 9b–d). This also strongly suggests that the basic principles of mammalian, including human, cortical development and evolution can be investigated in model systems such as the mouse [54, 55].

Mouse cortical interneurons (CINs) are derived from subcortical ganglionic eminences (GEs). a tSNE plot from scRNA-seq data (a total of 98,047 mouse cortical cells) was obtained using mouse E10.5 to the P4 cortex [306]. Individual cell types are labeled, adapted from Di Bella DJ et al., 2021 [306]. MGE-, POA-, and/or CGE-derived Dlx1+, and/or Dlx2+, Gad2+, Dlx5+, Sp8+, Sp9+, Nr2f2+, Lhx6+ CINs are clearly identified. Note that only a small number of IPCs for interneurons that express Egfr, Gsx2, Dlx1, Dlx2, and Gad2 are identified (arrows) adjacent to the bMIPC cluster that express Ascl1, Egfr, Olig1, and Olig2. Genetic labeling of the bMIPC experiments demonstrates that bMIPCs produce OBINs but not CINs. b It is well accepted that mouse CINs are derived from the MGE, POA, and CGE. c Mouse OBINs are derived from fRG cells in the developing MGE, LGE, cortex, and septum (SP). After birth, mouse OBINs are derived from B1 NSCs in the SVZ of the lateral ventricle. d Mouse cortical fRG cells first give rise to bMIPC, which then give rise to cortical APCs, OPCs, and OBIN-IPCs.

Virtually all human CINs are derived from subcortical ganglionic eminences (GEs), but not from the cortex. a tSNE plot from scRNA-seq data (a total of 57,868 human cortical cells) obtained using human GW18 (19,373 cells), GW22 (12,567 cells), GW23 (12,557 cells), and GW26 (13,371 cells) cortical tissue [88]. Individual cell types are labeled, adapted from Trevino et al., 2021 [88]. MGE-, POA-, and/or CGE-derived DLX1+, and/or DLX2+, GAD2+, DLX5+, SP8+, SP9+, NR2F2+, LHX6+ CINs are clearly identified. A small number of IPCs for interneurons express EGFR, GSX2, DLX1, DLX2, and GAD2 (arrows) are in the bMIPC cluster (ASCL1, EGFR, OLIG1, and OLIG2). This provides evidence that EGFR+, DLX1+, DLX2+, and GAD2+ IPCs are OBIN-IPCs, but not CIN-IPCs. Note that DLX5, SP8, and SP9 expression is not observed in the OBIN-IPC cluster, suggesting that very few immature OBINs are generated by GW23-26. b I propose that virtually all human CINs are derived from the MGE, POA, and CGE, similar to mice. c I propose that human OBINs are derived from fRG cells in the developing MGE, LGE, cortex, and septum (SP). After birth, human OBIN-IPCs are derived from B1 NSCs in the SVZ of the lateral ventricle. d Human cortical tRG cells first give rise to bMIPC, which then give rise to cortical APCs, OPCs, and OBIN-IPCs.
Human-specific Genes Alone May Not Be Essential for Most Cortical Expansion
The human cerebral cortex has 16.3 billion neurons, whereas the chimpanzee cerebral cortex has ~8 billion neurons. This discrepancy has led to extensive studies on human-specific genes over the past 10 years [104, 309,310,311], as these genes are believed to have significantly contributed to cortical expansion during human evolution. However, I propose that human-specific genes alone might not be fundamental for human cortical expansion.
The chimpanzee cerebral cortex has 8 billion neurons, whereas the macaque cortex has about 1.7 billion neurons. Human-specific genes cannot account for this. The expression of more than 20 human-specific protein-coding genes is enriched in human cortical progenitors [310]. Functional studies of these genes have revealed that many of them can expand cortical RG cells or PyN-IPCs. For example, overexpression of the human-specific gene ARHGAP11B in the embryonic mouse neocortex at E13.5 resulted in cortical folding at E18.5 [312]. Furthermore, the ARHGAP11B-transgenic marmoset fetal cortex exhibited surface folds at day 101 of the 150-day gestation, in contrast to the smooth surface of the normal control marmoset brain [313]. The authors suggest that ARHGAP11B plays a key role in human neocortex expansion. Thus, these human-specific genes may be important in the expansion of the human cortex, alone or in combination. Loss of function analysis in humans, human organoids, and/or experimental primates will be important to show which of these is critical for human cortical expansion. Perhaps some of these genes interact with the ERK-BMP7-GLI3R signaling pathway that I postulate is the core engine driving corticogenesis. Overall, I propose that those genes and signaling pathways that exhibit an evolutionary increase in expression in mammalian cortical RG cells or IPCs are important for cortical development, expansion, and evolution, such as phosphorylated ERK signaling and the BMP7 gene [54, 55].
Conclusions and Perspectives
In this review, based on our recent results and those of others, I began by describing the progression of mouse and human cortical RG cell lineages, emphasizing my perspective of their “slow and multistep” changes during the evolution of cortical neurogenesis. Second, I propose that the ERK-BMP7 signaling pathway in mammalian cortical RG cells plays a fundamental role in driving cortical development, expansion, and evolution. Finally, I propose that: (1) SHH signaling is not a key regulator of primate cortical expansion and folding; (2) human cortical radial glial cells do not generate neocortical interneurons; (3) human-specific genes may not be essential for most cortical expansion.
In 1962, Francis Crick said at the time he received the Nobel prize: “Politeness is the poison of all good collaboration in science. The soul of collaboration is perfect candor, and rudeness if need be. A good scientist values criticism almost higher than friendship: no, in science, criticism is the height and measure of friendship” [314]. In this review, I aim to provide clarity on what I believe is correct in the field. Of course, my proposals are hypotheses that should be critically evaluated and experimentally challenged. None-the-less, my fresh perspective merits serious consideration as it provides novel molecular insights into core mechanisms that may underlie how the cortex has expanded during evolution. It is based on evolutionarily old signaling pathways that are tinkered with to achieve increased cortical growth and the generation of a balanced set of cell types.
I have explained why I believe that human-specific genes may not be the central driver for cortical expansion. As shown in Fig. 1, it is clear that there must be a common rule that drives the expansion of the mammalian cortex and brain volume. Obviously, the expansion of the chimpanzee cerebral cortex compared with old-world monkeys has nothing to do with human-specific genes. As pointed out by Francois Jacob: “Evolution is a tinkerer. Evolution works on what already exists, either transforming a system to give it a new function or combining several systems to produce a more complex one” [315]. More and more human-specific genes are being identified. For example, around 900 genes with human-specific features were reported [316]. These human-specific genes may have a function in human brain evolution, but since they are human-specific they cannot be central to how the cortex has grown from rodents to monkeys and then to primates/humans. Other mechanisms must underlie this.
I have also explained why I believe that human cortical oRG cells cannot be versatile NSCs. Previous studies reported that human cortical oRG cells give rise not only to cortical PyNs but also to cortical OPCs and/or CINs in the second trimester [86, 89, 295,296,297]. It is believed that oRG cells in the OSVZ mainly produce PyNs for the upper cortical layers [56, 57, 60, 61, 98]. Again, as pointed out by Francois Jacob: “Evolution is far from perfection [315], and a new structural feature does not have to be optimal but must be ‘good enough’ to provide a survival advantage for the species” [162, 315]. I believe that the OSVZ cannot provide a niche for almost simultaneously producing cortical PyNs, OPCs, and CINs. In contrast, I believe that human cortical tRG-derived APCs and OPCs have great abilities to proliferate and migrate within the cortex. In other words, there is great evolutionary pressure for oRG cells to generate cortical PyNs, but it is not necessary for oRG cells to produce cortical OPCs and CINs.
As I proposed throughout the review, mammalian cortical evolutionary expansion is most likely driven by the ERK-BMP7-GLI3R signaling pathway in cortical RG cells, which participates in a positive feedback loop through antagonizing SHH signaling. Notably, this positive feedback loop can work like a tinkerer. Because ERK-BMP7-GLI3R signaling and SHH signaling mutually inhibit each other in cortical RG cells, they can drive either cortical evolutionary expansion or cortical evolutionary dwarfism.
And now, for my philosophical perspective. Earth is estimated to exist for at least 1 billion years; this would give us time for ERK-BMP7 signaling is continue driving our brains to evolve and generate a cortex with more and more neurons (Fig. 4). So, perhaps, in the future there will be more intelligent humans living on a peaceful blue planet.
References
Rakic P. A century of progress in corticoneurogenesis: From silver impregnation to genetic engineering. Cereb Cortex 2006, 16: i3–i17.
Hoch RV, Rubenstein JLR, Pleasure S. Genes and signaling events that establish regional patterning of the mammalian forebrain. Semin Cell Dev Biol 2009, 20: 378–386.
Fernandes M, Hébert JM. The ups and Downs of holoprosencephaly: Dorsal versus ventral patterning forces. Clin Genet 2008, 73: 413–423.
Hébert JM, Fishell G. The genetics of early telencephalon patterning: Some assembly required. Nat Rev Neurosci 2008, 9: 678–685.
Grove EA, Fukuchi-Shimogori T. Generating the cerebral cortical area map. Annu Rev Neurosci 2003, 26: 355–380.
Rash BG, Grove EA. Area and layer patterning in the developing cerebral cortex. Curr Opin Neurobiol 2006, 16: 25–34.
Cholfin JA, Rubenstein JLR. Frontal cortex subdivision patterning is coordinately regulated by Fgf8, Fgf17, and Emx2. J Comp Neurol 2008, 509: 144–155.
Fukuchi-Shimogori T, Grove EA. Neocortex patterning by the secreted signaling molecule FGF8. Science 2001, 294: 1071–1074.
Shimamura K, Rubenstein JL. Inductive interactions direct early regionalization of the mouse forebrain. Development 1997, 124: 2709–2718.
Crossley PH, Martinez S, Ohkubo Y, Rubenstein JL. Coordinate expression of Fgf8, Otx2, Bmp4, and Shh in the rostral prosencephalon during development of the telencephalic and optic vesicles. Neuroscience 2001, 108: 183–206.
Garel S, Huffman KJ, Rubenstein JLR. Molecular regionalization of the neocortex is disrupted in Fgf8 hypomorphic mutants. Development 2003, 130: 1903–1914.
Storm EE, Garel S, Borello U, Hebert JM, Martinez S, McConnell SK. Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 2006, 133: 1831–1844.
Ohkubo Y, Chiang C, Rubenstein JLR. Coordinate regulation and synergistic actions of BMP4, SHH and FGF8 in the rostral prosencephalon regulate morphogenesis of the telencephalic and optic vesicles. Neuroscience 2002, 111: 1–17.
Sur M, Rubenstein JLR. Patterning and plasticity of the cerebral cortex. Science 2005, 310: 805–810.
Abu-Khalil A, Fu L, Grove EA, Zecevic N, Geschwind DH. Wnt genes define distinct boundaries in the developing human brain: Implications for human forebrain patterning. J Comp Neurol 2004, 474: 276–288.
Jones WD, Guadiana SM, Grove EA. A model of neocortical area patterning in the lissencephalic mouse may hold for larger gyrencephalic brains. J Comp Neurol 2019, 527: 1461–1477.
Micali N, Ma S, Li M, Kim SK, Mato-Blanco X, Sindhu SK, et al. Molecular programs of regional specification and neural stem cell fate progression in macaque telencephalon. Science 2023, 382: eadf3786.
Braun E, Danan-Gotthold M, Borm LE, Lee KW, Vinsland E, Lönnerberg P, et al. Comprehensive cell atlas of the first-trimester developing human brain. Science 2023, 382: eadf1226.
Zeng B, Liu Z, Lu Y, Zhong S, Qin S, Huang L, et al. The single-cell and spatial transcriptional landscape of human gastrulation and early brain development. Cell Stem Cell 2023, 30: 851-866.e7.
Medina L, Abellán A. Development and evolution of the pallium. Semin Cell Dev Biol 2009, 20: 698–711.
Grant E, Hoerder-Suabedissen A, Molnár Z. Development of the corticothalamic projections. Front Neurosci 2012, 6: 53.
Backman M, Machon O, Mygland L, van den Bout CJ, Zhong W, Taketo MM, et al. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev Biol 2005, 279: 155–168.
Gunhaga L, Marklund M, Sjödal M, Hsieh JC, Jessell TM, Edlund T. Specification of dorsal telencephalic character by sequential Wnt and FGF signaling. Nat Neurosci 2003, 6: 701–707.
Machon O, Backman M, Machonova O, Kozmik Z, Vacik T, Andersen L, et al. A dynamic gradient of Wnt signaling controls initiation of neurogenesis in the mammalian cortex and cellular specification in the hippocampus. Dev Biol 2007, 311: 223–237.
Solberg N, Machon O, Krauss S. Effect of canonical Wnt inhibition in the neurogenic cortex, hippocampus, and premigratory dentate gyrus progenitor pool. Dev Dyn 2008, 237: 1799–1811.
Ille F, Atanasoski S, Falk S, Ittner LM, Märki D, Büchmann-Møller S, et al. Wnt/BMP signal integration regulates the balance between proliferation and differentiation of neuroepithelial cells in the dorsal spinal cord. Dev Biol 2007, 304: 394–408.
Hsu YC, Fuchs E. Building and maintaining the skin. Cold Spring Harb Perspect Biol 2022, 14: a040840.
Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327: 542–545.
Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 2002, 297: 365–369.
Shimogori T, Banuchi V, Ng HY, Strauss JB, Grove EA. Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development 2004, 131: 5639–5647.
Saulnier A, Keruzore M, De Clercq S, Bar I, Moers V, Magnani D, et al. The doublesex homolog Dmrt5 is required for the development of the caudomedial cerebral cortex in mammals. Cereb Cortex 2013, 23: 2552–2567.
Caronia-Brown G, Yoshida M, Gulden F, Assimacopoulos S, Grove EA. The cortical hem regulates the size and patterning of neocortex. Development 2014, 141: 2855–2865.
Theil T, Aydin S, Koch S, Grotewold L, Rüther U. Wnt and Bmp signalling cooperatively regulate graded Emx2 expression in the dorsal telencephalon. Development 2002, 129: 3045–3054.
Chou SJ, Tole S. Lhx2, an evolutionarily conserved, multifunctional regulator of forebrain development. Brain Res 2019, 1705: 1–14.
Aoto K, Nishimura T, Eto K, Motoyama J. Mouse GLI3 regulates Fgf8 expression and apoptosis in the developing neural tube, face, and limb bud. Dev Biol 2002, 251: 320–332.
Rash BG, Grove EA. Patterning the dorsal telencephalon: A role for sonic hedgehog? J Neurosci 2007, 27: 11595–11603.
Kuschel S, Rüther U, Theil T. A disrupted balance between Bmp/Wnt and Fgf signaling underlies the ventralization of the Gli3 mutant telencephalon. Dev Biol 2003, 260: 484–495.
Theil T, Alvarez-Bolado G, Walter A, Rüther U. Gli3 is required for Emx gene expression during dorsal telencephalon development. Development 1999, 126: 3561–3571.
Tole S, Ragsdale CW, Grove EA. Dorsoventral patterning of the telencephalon is disrupted in the mouse mutant extra-toes(J). Dev Biol 2000, 217: 254–265.
Hasenpusch-Theil K, Magnani D, Amaniti EM, Han L, Armstrong D, Theil T. Transcriptional analysis of Gli3 mutants identifies Wnt target genes in the developing hippocampus. Cereb Cortex 2012, 22: 2878–2893.
Huang X, Litingtung Y, Chiang C. Ectopic sonic hedgehog signaling impairs telencephalic dorsal midline development: Implication for human holoprosencephaly. Hum Mol Genet 2007, 16: 1454–1468.
Huang X, Litingtung Y, Chiang C. Region-specific requirement for cholesterol modification of sonic hedgehog in patterning the telencephalon and spinal cord. Development 2007, 134: 2095–2105.
Herculano-Houzel S, Avelino-de-Souza K, Neves K, Porfírio J, Messeder D, Mattos Feijó L, et al. The elephant brain in numbers. Front Neuroanat 2014, 8: 46.
Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, Leite REP, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol 2009, 513: 532–541.
Herculano-Houzel S, Catania K, Manger PR, Kaas JH. Mammalian brains are made of these: A dataset of the numbers and densities of neuronal and nonneuronal cells in the brain of glires, Primates, Scandentia, eulipotyphlans, afrotherians and artiodactyls, and their relationship with body mass. Brain Behav Evol 2015, 86: 145–163.
Sousa AMM, Meyer KA, Santpere G, Gulden FO, Sestan N. Evolution of the human nervous system function, structure, and development. Cell 2017, 170: 226–247.
Anthony TE, Klein C, Fishell G, Heintz N. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron 2004, 41: 881–890.
Gao P, Postiglione MP, Krieger TG, Hernandez L, Wang C, Han Z, et al. Deterministic progenitor behavior and unitary production of neurons in the neocortex. Cell 2014, 159: 775–788.
Guo C, Eckler MJ, McKenna WL, McKinsey GL, Rubenstein JLR, Chen B. Fezf2 expression identifies a multipotent progenitor for neocortical projection neurons, astrocytes, and oligodendrocytes. Neuron 2013, 80: 1167–1174.
Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 2009, 32: 149–184.
Kwan KY, Sestan N, Anton ES. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 2012, 139: 1535–1546.
Lin Y, Yang J, Shen Z, Ma J, Simons BD, Shi SH. Behavior and lineage progression of neural progenitors in the mammalian cortex. Curr Opin Neurobiol 2021, 66: 144–157.
Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature 2001, 409: 714–720.
Sun M, Gao Y, Li Z, Yang L, Liu G, Xu Z, et al. ERK signaling expands mammalian cortical radial glial cells and extends the neurogenic period. Proc Natl Acad Sci U S A 2024, 121: e2314802121.
Li Z, Liu G, Yang L, Sun M, Zhang Z, Xu Z, et al. BMP7 expression in mammalian cortical radial glial cells increases the length of the neurogenic period. Protein Cell 2024, 15: 21–35.
Hansen DV, Lui JH, Parker PRL, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 2010, 464: 554–561.
Fietz SA, Kelava I, Vogt J, Wilsch-Bräuninger M, Stenzel D, Fish JL, et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci 2010, 13: 690–699.
Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, et al. Molecular identity of human outer radial glia during cortical development. Cell 2015, 163: 55–67.
Nowakowski TJ, Pollen AA, Sandoval-Espinosa C, Kriegstein AR. Transformation of the radial Glia scaffold demarcates two stages of human cerebral cortex development. Neuron 2016, 91: 1219–1227.
Yang L, Li Z, Liu G, Li X, Yang Z. Developmental origins of human cortical oligodendrocytes and astrocytes. Neurosci Bull 2022, 38: 47–68.
Reillo I, de Juan Romero C, García-Cabezas MÁ, Borrell V. A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb Cortex 2011, 21: 1674–1694.
LaMonica BE, Lui JH, Hansen DV, Kriegstein AR. Mitotic spindle orientation predicts outer radial glial cell generation in human neocortex. Nat Commun 2013, 4: 1665.
Baburamani AA, Vontell RT, Uus A, Pietsch M, Patkee PA, Wyatt-Ashmead J, et al. Assessment of radial glia in the frontal lobe of fetuses with Down syndrome. Acta Neuropathol Commun 2020, 8: 141.
Smart IHM, Dehay C, Giroud P, Berland M, Kennedy H. Unique morphological features of the proliferative zones and postmitotic compartments of the neural epithelium giving rise to striate and extrastriate cortex in the monkey. Cereb Cortex 2002, 12: 37–53.
Dehay C, Kennedy H, Kosik KS. The outer subventricular zone and primate-specific cortical complexification. Neuron 2015, 85: 683–694.
Betizeau M, Cortay V, Patti D, Pfister S, Gautier E, Bellemin-Ménard A, et al. Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron 2013, 80: 442–457.
Leone DP, Srinivasan K, Chen B, Alcamo E, McConnell SK. The determination of projection neuron identity in the developing cerebral cortex. Curr Opin Neurobiol 2008, 18: 28–35.
Florio M, Huttner WB. Neural progenitors, neurogenesis and the evolution of the neocortex. Development 2014, 141: 2182–2194.
Rakic P. Neurogenesis in adult primate neocortex: An evaluation of the evidence. Nat Rev Neurosci 2002, 3: 65–71.
Merkle FT, Mirzadeh Z, Alvarez-Buylla A. Mosaic organization of neural stem cells in the adult brain. Science 2007, 317: 381–384.
Ventura RE, Goldman JE. Dorsal radial glia generate olfactory bulb interneurons in the postnatal murine brain. J Neurosci 2007, 27: 4297–4302.
Young KM, Fogarty M, Kessaris N, Richardson WD. Subventricular zone stem cells are heterogeneous with respect to their embryonic origins and neurogenic fates in the adult olfactory bulb. J Neurosci 2007, 27: 8286–8296.
Kohwi M, Petryniak MA, Long JE, Ekker M, Obata K, Yanagawa Y, et al. A subpopulation of olfactory bulb GABAergic interneurons is derived from Emx1- and Dlx5/6-expressing progenitors. J Neurosci 2007, 27: 6878–6891.
Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci 2006, 9: 173–179.
Cai Y, Zhang Y, Shen Q, Rubenstein JLR, Yang Z. A subpopulation of individual neural progenitors in the mammalian dorsal pallium generates both projection neurons and interneurons in vitro. Stem Cells 2013, 31: 1193–1201.
Fuentealba LC, Rompani SB, Parraguez JI, Obernier K, Romero R, Cepko CL, et al. Embryonic origin of postnatal neural stem cells. Cell 2015, 161: 1644–1655.
Gao P, Sultan KT, Zhang XJ, Shi SH. Lineage-dependent circuit assembly in the neocortex. Development 2013, 140: 2645–2655.
Eckler MJ, Nguyen TD, McKenna WL, Fastow BL, Guo C, Rubenstein JLR, et al. Cux2-positive radial glial cells generate diverse subtypes of neocortical projection neurons and macroglia. Neuron 2015, 86: 1100–1108.
Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 2004, 7: 136–144.
Li X, Liu G, Yang L, Li Z, Zhang Z, Xu Z, et al. Decoding cortical glial cell development. Neurosci Bull 2021, 37: 440–460.
Huang H, Rubenstein JL, Qiu M. Cracking the codes of cortical glial progenitors: Evidence for the common lineage of astrocytes and oligodendrocytes. Neurosci Bull 2021, 37: 437–439.
Huang H, He W, Tang T, Qiu M. Immunological markers for central nervous system Glia. Neurosci Bull 2023, 39: 379–392.
Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature 1983, 303: 390–396.
Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature 2010, 468: 214–222.
Zhang Y, Liu G, Guo T, Liang XG, Du H, Yang L, et al. Cortical neural stem cell lineage progression is regulated by extrinsic signaling molecule sonic hedgehog. Cell Rep 2020, 30: 4490-4504.e4.
Huang W, Bhaduri A, Velmeshev D, Wang S, Wang L, Rottkamp CA, et al. Origins and proliferative states of human oligodendrocyte precursor cells. Cell 2020, 182: 594-608.e11.
Fu Y, Yang M, Yu H, Wang Y, Wu X, Yong J, et al. Heterogeneity of glial progenitor cells during the neurogenesis-to-gliogenesis switch in the developing human cerebral cortex. Cell Rep 2021, 34: 108788.
Trevino AE, Müller F, Andersen J, Sundaram L, Kathiria A, Shcherbina A, et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 2021, 184: 5053-5069.e23.
Liu DD, He JQ, Sinha R, Eastman AE, Toland AM, Morri M, et al. Purification and characterization of human neural stem and progenitor cells. Cell 2023, 186: 1179-1194.e15.
Weng Q, Wang J, Wang J, He D, Cheng Z, Zhang F, et al. Single-cell transcriptomics uncovers glial progenitor diversity and cell fate determinants during development and gliomagenesis. Cell Stem Cell 2019, 24: 707-723.e8.
Shen Z, Lin Y, Yang J, Jörg DJ, Peng Y, Zhang X, et al. Distinct progenitor behavior underlying neocortical gliogenesis related to tumorigenesis. Cell Rep 2021, 34: 108853.
Moreau MX, Saillour Y, Cwetsch AW, Pierani A, Causeret F. Single-cell transcriptomics of the early developing mouse cerebral cortex disentangle the spatial and temporal components of neuronal fate acquisition. Development 2021, 148: dev197962.
Faedo A, Tomassy GS, Ruan Y, Teichmann H, Krauss S, Pleasure SJ, et al. COUP-TFI coordinates cortical patterning, neurogenesis, and laminar fate and modulates MAPK/ERK, AKT, and ß-catenin signaling. Cereb Cortex 2008, 18: 2117–2131.
Ypsilanti AR, Pattabiraman K, Catta-Preta R, Golonzhka O, Lindtner S, Tang K, et al. Transcriptional network orchestrating regional patterning of cortical progenitors. Proc Natl Acad Sci U S A 2021, 118: e2024795118.
Borello U, Madhavan M, Vilinsky I, Faedo A, Pierani A, Rubenstein J, et al. Sp8 and COUP-TF1 reciprocally regulate patterning and fgf signaling in cortical progenitors. Cereb Cortex 2014, 24: 1409–1421.
O’Leary DDM, Chou SJ, Sahara S. Area patterning of the mammalian cortex. Neuron 2007, 56: 252–269.
Du H, Wang Z, Guo R, Yang L, Liu G, Zhang Z, et al. Transcription factors Bcl11a and Bcl11b are required for the production and differentiation of cortical projection neurons. Cereb Cortex 2022, 32: 3611–3632.
Lui JH, Hansen DV, Kriegstein AR. Development and evolution of the human neocortex. Cell 2011, 146: 18–36.
Bystron I, Blakemore C, Rakic P. Development of the human cerebral cortex: Boulder Committee revisited. Nat Rev Neurosci 2008, 9: 110–122.
Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 2017, 358: 1318–1323.
Rakic P. Elusive radial glial cells: Historical and evolutionary perspective. Glia 2003, 43: 19–32.
Molnár Z, Clowry GJ, Šestan N, Alzu’bi A, Bakken T, Hevner RF, et al. New insights into the development of the human cerebral cortex. J Anat 2019, 235: 432–451.
Cárdenas A, Borrell V. Molecular and cellular evolution of corticogenesis in amniotes. Cell Mol Life Sci 2020, 77: 1435–1460.
Pollen AA, Kilik U, Lowe CB, Camp JG. Human-specific genetics: New tools to explore the molecular and cellular basis of human evolution. Nat Rev Genet 2023, 24: 687–711.
Merkle FT, Tramontin AD, García-Verdugo JM, Alvarez-Buylla A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci U S A 2004, 101: 17528–17532.
Lim DA, Alvarez-Buylla A. The adult ventricular-subventricular zone (V-SVZ) and olfactory bulb (OB) neurogenesis. Cold Spring Harb Perspect Biol 2016, 8: a018820.
Alvarez-Buylla A, Kohwi M, Nguyen TM, Merkle FT. The heterogeneity of adult neural stem cells and the emerging complexity of their niche. Cold Spring Harb Symp Quant Biol 2008, 73: 357–365.
Zhou X, Liu F, Tian M, Xu Z, Liang Q, Wang C, et al. Transcription factors COUP-TFI and COUP-TFII are required for the production of granule cells in the mouse olfactory bulb. Development 2015, 142: 1593–1605.
Obernier K, Alvarez-Buylla A. Neural stem cells: Origin, heterogeneity and regulation in the adult mammalian brain. Development 2019, 146: dev156059.
Ma T, Wang C, Wang L, Zhou X, Tian M, Zhang Q, et al. Subcortical origins of human and monkey neocortical interneurons. Nat Neurosci 2013, 16: 1588–1597.
Wang C, Liu F, Liu YY, Zhao CH, You Y, Wang L, et al. Identification and characterization of neuroblasts in the subventricular zone and rostral migratory stream of the adult human brain. Cell Res 2011, 21: 1534–1550.
Sanai N, Nguyen T, Ihrie RA, Mirzadeh Z, Tsai HH, Wong M, et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature 2011, 478: 382–386.
Wang C, You Y, Qi D, Zhou X, Wang L, Wei S, et al. Human and monkey striatal interneurons are derived from the medial ganglionic eminence but not from the adult subventricular zone. J Neurosci 2014, 34: 10906–10923.
Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555: 377–381.
Nascimento MA, Biagiotti S, Herranz-Pérez V, Santiago S, Bueno R, Ye CJ, et al. Protracted neuronal recruitment in the temporal lobes of young children. Nature 2024, 626: 1056–1065.
Sorrells SF, Paredes MF, Zhang Z, Kang G, Pastor-Alonso O, Biagiotti S, et al. Positive controls in adults and children support that very few, if any, new neurons are born in the adult human hippocampus. J Neurosci 2021, 41: 2554–2565.
Paredes MF, James D, Gil-Perotin S, Kim H, Cotter JA, Ng C, et al. Extensive migration of young neurons into the infant human frontal lobe. Science 2016, 354: aaf7073.
Paredes MF, Sorrells SF, Garcia-Verdugo JM, Alvarez-Buylla A. Brain size and limits to adult neurogenesis. J Comp Neurol 2016, 524: 646–664.
Girskis KM, Stergachis AB, DeGennaro EM, Doan RN, Qian X, Johnson MB, et al. Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Neuron 2021, 109: 3239-3251.e7.
Lewitus E, Kelava I, Kalinka AT, Tomancak P, Huttner WB. An adaptive threshold in mammalian neocortical evolution. PLoS Biol 2014, 12: e1002000.
Lin L, Zhao J, Kubota N, Li Z, Lam YL, Nguyen LP, et al. 2024 Epistatic interactions between NMD and TRP53 control progenitor cell maintenance and brain size. Neuron 2024, 112: 2157–2176 e2112.
Arai Y, Pulvers JN, Haffner C, Schilling B, Nüsslein I, Calegari F, et al. Neural stem and progenitor cells shorten S-phase on commitment to neuron production. Nat Commun 2011, 2: 154.
Takahashi T, Nowakowski RS Jr, Caviness VS. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci 1995, 15: 6046–6057.
Shen Q, Wang Y, Dimos JT, Fasano CA, Phoenix TN, Lemischka IR, et al. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci 2006, 9: 743–751.
Kornack DR, Rakic P. Changes in cell-cycle kinetics during the development and evolution of primate neocortex. Proc Natl Acad Sci U S A 1998, 95: 1242–1246.
Stepien BK, Vaid S, Huttner WB. Length of the neurogenic period-a key determinant for the generation of upper-layer neurons during neocortex development and evolution. Front Cell Dev Biol 2021, 9: 676911.
Dehay C, Kennedy H. Cell-cycle control and cortical development. Nat Rev Neurosci 2007, 8: 438–450.
Stepien BK, Naumann R, Holtz A, Helppi J, Huttner WB, Vaid S. Lengthening neurogenic period during neocortical development causes a hallmark of neocortex expansion. Curr Biol 2020, 30: 4227-4237.e5.
Hu X, Eastman AE, Guo S. Cell cycle dynamics in the reprogramming of cellular identity. FEBS Lett 2019, 593: 2840–2852.
Soufi A, Dalton S. Cycling through developmental decisions: How cell cycle dynamics control pluripotency, differentiation and reprogramming. Development 2016, 143: 4301–4311.
Guo T, Liu G, Du H, Wen Y, Wei S, Li Z, et al. Dlx1/2 are central and essential components in the transcriptional code for generating olfactory bulb interneurons. Cereb Cortex 2019, 29: 4831–4849.
Gaiano N, Nye JS, Fishell G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 2000, 26: 395–404.
Shimojo H, Ohtsuka T, Kageyama R. Oscillations in Notch signaling regulate maintenance of neural progenitors. Neuron 2008, 58: 52–64.
Harris L, Guillemot F. HES1, two programs: Promoting the quiescence and proliferation of adult neural stem cells. Genes Dev 2019, 33: 479–481.
Guo R, Han D, Song X, Gao Y, Li Z, Li X, et al. Context-dependent regulation of Notch signaling in glial development and tumorigenesis. Sci Adv 2023, 9: eadi2167.
Kang W, Wong LC, Shi SH, Hébert JM. The transition from radial glial to intermediate progenitor cell is inhibited by FGF signaling during corticogenesis. J Neurosci 2009, 29: 14571–14580.
Rash BG, Lim HD, Breunig JJ, Vaccarino FM. FGF signaling expands embryonic cortical surface area by regulating Notch-dependent neurogenesis. J Neurosci 2011, 31: 15604–15617.
Imamura O, Pagès G, Pouysségur J, Endo S, Takishima K. ERK1 and ERK2 are required for radial glial maintenance and cortical lamination. Genes Cells 2010, 15: 1072–1088.
Lavoie H, Gagnon J, Therrien M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol 2020, 21: 607–632.
Li S, Mattar P, Dixit R, Lawn SO, Wilkinson G, Kinch C, et al. RAS/ERK signaling controls proneural genetic programs in cortical development and gliomagenesis. J Neurosci 2014, 34: 2169–2190.
Faedo A, Borello U, Rubenstein JLR. Repression of Fgf signaling by sprouty1-2 regulates cortical patterning in two distinct regions and times. J Neurosci 2010, 30: 4015–4023.
Hasenpusch-Theil K, West S, Kelman A, Kozic Z, Horrocks S, McMahon AP, et al. Gli3 controls the onset of cortical neurogenesis by regulating the radial glial cell cycle through Cdk6 expression. Development 2018, 145: dev163147.
Komada M, Saitsu H, Kinboshi M, Miura T, Shiota K, Ishibashi M. Hedgehog signaling is involved in development of the neocortex. Development 2008, 135: 2717–2727.
Komada M, Iguchi T, Takeda T, Ishibashi M, Sato M. Smoothened controls cyclin D2 expression and regulates the generation of intermediate progenitors in the developing cortex. Neurosci Lett 2013, 547: 87–91.
Heng X, Guo Q, Leung AW, Li JY. Analogous mechanism regulating formation of neocortical basal radial glia and cerebellar Bergmann glia. Elife 2017, 6: 23253.
Lui JH, Nowakowski TJ, Pollen AA, Javaherian A, Kriegstein AR, Oldham MC. Radial glia require PDGFD-PDGFRβ signalling in human but not mouse neocortex. Nature 2014, 515: 264–268.
Johnson MB, Wang PP, Atabay KD, Murphy EA, Doan RN, Hecht JL, et al. Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat Neurosci 2015, 18: 637–646.
Delgado AC, Maldonado-Soto AR, Silva-Vargas V, Mizrak D, von Känel T, Tan KR, et al. Release of stem cells from quiescence reveals gliogenic domains in the adult mouse brain. Science 2021, 372: 1205–1209.
Klein RS, Rubin JB, Gibson HD, DeHaan EN, Alvarez-Hernandez X, Segal RA, et al. SDF-1 alpha induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development 2001, 128: 1971–1981.
Walenkamp AME, Lapa C, Herrmann K, Wester HJ. CXCR4 ligands: The next big hit? J Nucl Med 2017, 58: 77S-82S.
Talley MJ, Nardini D, Qin S, Prada CE, Ehrman LA, Waclaw RR. A role for sustained MAPK activity in the mouse ventral telencephalon. Dev Biol 2021, 476: 137–147.
Winkler CC, Yabut OR, Fregoso SP, Gomez HG, Dwyer BE, Pleasure SJ, et al. The dorsal wave of neocortical oligodendrogenesis begins embryonically and requires multiple sources of sonic hedgehog. J Neurosci 2018, 38: 5237–5250.
Zhang X, Mennicke CV, Xiao G, Beattie R, Haider MA, Hippenmeyer S, et al. Clonal analysis of gliogenesis in the cerebral cortex reveals stochastic expansion of Glia and cell autonomous responses to egfr dosage. Cells 2020, 9: 2662.
Genander M, Cook PJ, Ramsköld D, Keyes BE, Mertz AF, Sandberg R, et al. BMP signaling and its pSMAD1/5 target genes differentially regulate hair follicle stem cell lineages. Cell Stem Cell 2014, 15: 619–633.
Adam RC, Yang H, Ge Y, Lien WH, Wang P, Zhao Y, et al. Temporal layering of signaling effectors drives chromatin remodeling during hair follicle stem cell lineage progression. Cell Stem Cell 2018, 22: 398-413.e7.
Hara T, Tanegashima K. CXCL14 antagonizes the CXCL12-CXCR4 signaling axis. Biomol Concepts 2014, 5: 167–173.
Martynoga B, Mateo JL, Zhou B, Andersen J, Achimastou A, Urbán N, et al. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev 2013, 27: 1769–1786.
Sun Y, Hu J, Zhou L, Pollard SM, Smith A. Interplay between FGF2 and BMP controls the self-renewal, dormancy and differentiation of rat neural stem cells. J Cell Sci 2011, 124: 1867–1877.
Kretzschmar M, Doody J, Massagué J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 1997, 389: 618–622.
Pera EM, Ikeda A, Eivers E, De Robertis EM. Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev 2003, 17: 3023–3028.
Rakic P. Specification of cerebral cortical areas. Science 1988, 241: 170–176.
Rakic P. A small step for the cell, a giant leap for mankind: A hypothesis of neocortical expansion during evolution. Trends Neurosci 1995, 18: 383–388.
Rakic P. Evolution of the neocortex: A perspective from developmental biology. Nat Rev Neurosci 2009, 10: 724–735.
Picco N, García-Moreno F, Maini PK, Woolley TE, Molnár Z. Mathematical modeling of cortical neurogenesis reveals that the founder population does not necessarily scale with neurogenic output. Cereb Cortex 2018, 28: 2540–2550.
Galakhova AA, Hunt S, Wilbers R, Heyer DB, de Kock CPJ, Mansvelder HD, et al. Evolution of cortical neurons supporting human cognition. Trends Cogn Sci 2022, 26: 909–922.
DeFelipe J, Alonso-Nanclares L, Arellano JI. Microstructure of the neocortex: Comparative aspects. J Neurocytol 2002, 31: 299–316.
Lüsebrink F, Wollrab A, Speck O. Cortical thickness determination of the human brain using high resolution 3T and 7T MRI data. NeuroImage 2013, 70: 122–131.
Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A 2000, 97: 11050–11055.
Espinós A, Fernández-Ortuño E, Negri E, Borrell V. Evolution of genetic mechanisms regulating cortical neurogenesis. Dev Neurobiol 2022, 82: 428–453.
Kolk SM, Rakic P. Development of prefrontal cortex. Neuropsychopharmacology 2022, 47: 41–57.
Bizzotto S, Walsh CA. Genetic mosaicism in the human brain: From lineage tracing to neuropsychiatric disorders. Nat Rev Neurosci 2022, 23: 275–286.
Luria V, Ma S, Shibata M, Pattabiraman K, Sestan N. Molecular and cellular mechanisms of human cortical connectivity. Curr Opin Neurobiol 2023, 80: 102699.
Taverna E, Götz M, Huttner WB. The cell biology of neurogenesis: Toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol 2014, 30: 465–502.
Dehay C, Huttner WB. Development and evolution of the primate neocortex from a progenitor cell perspective. Development 2024, 151: dev199797.
Huttner WB, Heide M, Mora-Bermúdez F, Namba T. Neocortical neurogenesis in development and evolution-Human-specific features. J Comp Neurol 2024, 532: e25576.
Shao Y, Zhou L, Li F, Zhao L, Zhang BL, Shao F, et al. Phylogenomic analyses provide insights into primate evolution. Science 2023, 380: 913–924.
Kovacević N, Henderson JT, Chan E, Lifshitz N, Bishop J, Evans AC, et al. A three-dimensional MRI atlas of the mouse brain with estimates of the average and variability. Cereb Cortex 2005, 15: 639–645.
Sawada K, Horiuchi-Hirose M, Saito S, Aoki I. MRI-based morphometric characterizations of sexual dimorphism of the cerebrum of ferrets (Mustela putorius). NeuroImage 2013, 83: 294–306.
Mira H, Andreu Z, Suh H, Lie DC, Jessberger S, Consiglio A, et al. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem Cell 2010, 7: 78–89.
Urbán N, Guillemot F. Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front Cell Neurosci 2014, 8: 396.
Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG, et al. Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev 2001, 15: 2094–2110.
Lim DA, Tramontin AD, Trevejo JM, Herrera DG, García-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron 2000, 28: 713–726.
Echevarría D, Vieira C, Gimeno L, Martínez S. Neuroepithelial secondary organizers and cell fate specification in the developing brain. Brain Res Brain Res Rev 2003, 43: 179–191.
Hoch RV, Clarke JA, Rubenstein JL. Fgf signaling controls the telencephalic distribution of Fgf-expressing progenitors generated in the rostral patterning center. Neural Dev 2015, 10: 8.
Wilson SW, Houart C. Early steps in the development of the forebrain. Dev Cell 2004, 6: 167–181.
Pani AM, Mullarkey EE, Aronowicz J, Assimacopoulos S, Grove EA, Lowe CJ. Ancient deuterostome origins of vertebrate brain signalling centres. Nature 2012, 483: 289–294.
Borello U, Cobos I, Long JE, McWhirter JR, Murre C, Rubenstein JL. FGF15 promotes neurogenesis and opposes FGF8 function during neocortical development. Neural Dev 2008, 3: 17.
Segklia A, Seuntjens E, Elkouris M, Tsalavos S, Stappers E, Mitsiadis TA, et al. Bmp7 regulates the survival, proliferation, and neurogenic properties of neural progenitor cells during corticogenesis in the mouse. PLoS One 2012, 7: e34088.
Guillemot F, Zimmer C. From cradle to grave: The multiple roles of fibroblast growth factors in neural development. Neuron 2011, 71: 574–588.
Iwata T, Hevner RF. Fibroblast growth factor signaling in development of the cerebral cortex. Dev Growth Differ 2009, 51: 299–323.
Mason I. Initiation to end point: The multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci 2007, 8: 583–596.
Ünal EB, Uhlitz F, Blüthgen N. A compendium of ERK targets. FEBS Lett 2017, 591: 2607–2615.
Maik-Rachline G, Hacohen-Lev-Ran A, Seger R. Nuclear ERK: Mechanism of translocation, substrates, and role in cancer. Int J Mol Sci 2019, 20: 1194.
Hu S, Xie Z, Onishi A, Yu X, Jiang L, Lin J, et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 2009, 139: 610–622.
Göke J, Chan YS, Yan J, Vingron M, Ng HH. Genome-wide kinase-chromatin interactions reveal the regulatory network of ERK signaling in human embryonic stem cells. Mol Cell 2013, 50: 844–855.
Tee WW, Shen SS, Oksuz O, Narendra V, Reinberg D. Erk1/2 activity promotes chromatin features and RNAPII phosphorylation at developmental promoters in mouse ESCs. Cell 2014, 156: 678–690.
Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009, 139: 573–586.
Knowles SJ, Stafford AM, Zaman T, Angara K, Williams MR, Newbern JM, et al. Distinct hyperactive RAS/MAPK alleles converge on common GABAergic interneuron core programs. Development 2023, 150: dev201371.
Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, et al. MEK is a key regulator of gliogenesis in the developing brain. Neuron 2012, 75: 1035–1050.
Douceau S, Deutsch Guerrero T, Ferent J. Establishing hedgehog gradients during neural development. Cells 2023, 12: 225.
Yabut OR, Ng HX, Yoon K, Arela JC, Ngo T, Pleasure SJ. The neocortical progenitor specification program is established through combined modulation of SHH and FGF signaling. J Neurosci 2020, 40: 6872–6887.
Zhuang XL, Zhang JJ, Shao Y, Ye Y, Chen CY, Zhou L, et al. Integrative omics reveals rapidly evolving regulatory sequences driving primate brain evolution. Mol Biol Evol 2023, 40: msad173.
Kelava I, Lewitus E, Huttner WB. The secondary loss of gyrencephaly as an example of evolutionary phenotypical reversal. Front Neuroanat 2013, 7: 16.
Ornitz DM, Legeai-Mallet L. Achondroplasia: Development, pathogenesis, and therapy. Dev Dyn 2017, 246: 291–309.
Hevner RF. The cerebral cortex malformation in thanatophoric dysplasia: Neuropathology and pathogenesis. Acta Neuropathol 2005, 110: 208–221.
Alzu’bi A, Lindsay SJ, Harkin LF, McIntyre J, Lisgo SN, Clowry GJ. The transcription factors COUP-TFI and COUP-TFII have distinct roles in arealisation and GABAergic interneuron specification in the early human fetal telencephalon. Cereb Cortex 2017, 27: 4971–4987.
de Juan Romero C, Bruder C, Tomasello U, Sanz-Anquela JM, Borrell V. Discrete domains of gene expression in germinal layers distinguish the development of gyrencephaly. EMBO J 2015, 34: 1859–1874.
Fukuchi-Shimogori T, Grove EA. Emx2 patterns the neocortex by regulating FGF positional signaling. Nat Neurosci 2003, 6: 825–831.
Thomson RE, Pellicano F, Iwata T. Fibroblast growth factor receptor 3 kinase domain mutation increases cortical progenitor proliferation via mitogen-activated protein kinase activation. J Neurochem 2007, 100: 1565–1578.
Yasoda A, Komatsu Y, Chusho H, Miyazawa T, Ozasa A, Miura M, et al. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med 2004, 10: 80–86.
Lin T, Sandusky SB, Xue H, Fishbein KW, Spencer RG, Rao MS, et al. A central nervous system specific mouse model for thanatophoric dysplasia type II. Hum Mol Genet 2003, 12: 2863–2871.
Iwata T, Chen L, Li C, Ovchinnikov DA, Behringer RR, Francomano CA, et al. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum Mol Genet 2000, 9: 1603–1613.
Inglis-Broadgate SL, Thomson RE, Pellicano F, Tartaglia MA, Pontikis CC, Cooper JD, et al. FGFR3 regulates brain size by controlling progenitor cell proliferation and apoptosis during embryonic development. Dev Biol 2005, 279: 73–85.
Matsushita T, Wilcox WR, Chan YY, Kawanami A, Bükülmez H, Balmes G, et al. FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum Mol Genet 2009, 18: 227–240.
Li C, Chen L, Iwata T, Kitagawa M, Fu XY, Deng CX. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet 1999, 8: 35–44.
Wyatt AW, Osborne RJ, Stewart H, Ragge NK. Bone morphogenetic protein 7 (BMP7) mutations are associated with variable ocular, brain, ear, palate, and skeletal anomalies. Hum Mutat 2010, 31: 781–787.
Wang L, Park JY, Liu F, Olesen KM, Hou S, Peng JC, et al. A kinase-independent function of cyclin-dependent kinase 6 promotes outer radial glia expansion and neocortical folding. Proc Natl Acad Sci U S A 2022, 119: e2206147119.
Wang L, Hou S, Han YG. Hedgehog signaling promotes basal progenitor expansion and the growth and folding of the neocortex. Nat Neurosci 2016, 19: 888–896.
Akula SK, Marciano JH, Lim Y, Exposito-Alonso D, Hylton NK, Hwang GH, et al. TMEM161B regulates cerebral cortical gyration, Sonic Hedgehog signaling, and ciliary structure in the developing central nervous system. Proc Natl Acad Sci U S A 2023, 120: e2209964120.
Akula SK, Exposito-Alonso D, Walsh CA. Shaping the brain: The emergence of cortical structure and folding. Dev Cell 2023, 58: 2836–2849.
Coulter ME, Dorobantu CM, Lodewijk GA, Delalande F, Cianferani S, Ganesh VS, et al. The ESCRT-III protein CHMP1A mediates secretion of sonic hedgehog on a distinctive subtype of extracellular vesicles. Cell Rep 2018, 24: 973-986.e8.
Hou S, Ho WL, Wang L, Kuo B, Park JY, Han YG. Biphasic roles of hedgehog signaling in the production and self-renewal of outer radial Glia in the ferret cerebral cortex. Cereb Cortex 2021, 31: 4730–4741.
Han YG. Sonic hedgehog signaling: A conserved mechanism for the expansion of outer radial glia and intermediate progenitor cells and for the growth and folding of the neocortex. Neurogenesis 2016, 3: e1242957.
Moffat A, Schuurmans C 2023 The control of cortical folding: Multiple mechanisms, multiple models. Neuroscientist: 10738584231190839.
Penisson M, Ladewig J, Belvindrah R, Francis F. Genes and mechanisms involved in the generation and amplification of basal radial glial cells. Front Cell Neurosci 2019, 13: 381.
Shimada IS, Somatilaka BN, Hwang SH, Anderson AG, Shelton JM, Rajaram V, et al. Derepression of sonic hedgehog signaling upon Gpr161 deletion unravels forebrain and ventricular abnormalities. Dev Biol 2019, 450: 47–62.
Matsumoto N, Tanaka S, Horiike T, Shinmyo Y, Kawasaki H. A discrete subtype of neural progenitor crucial for cortical folding in the gyrencephalic mammalian brain. Elife 2020, 9: e54873.
Cai E, Barba MG, Ge X. Hedgehog signaling in cortical development. Cells 2023, 13: 21.
Ohtsuka T, Kageyama R. Dual activation of Shh and Notch signaling induces dramatic enlargement of neocortical surface area. Neurosci Res 2022, 176: 18–30.
Dave RK, Ellis T, Toumpas MC, Robson JP, Julian E, Adolphe C, et al. Sonic hedgehog and Notch signaling can cooperate to regulate neurogenic divisions of neocortical progenitors. PLoS One 2011, 6: e14680.
Fuccillo M, Joyner AL, Fishell G. Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci 2006, 7: 772–783.
Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22: 103–114.
Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: The Current consensus. Acta Neuropathol 2012, 123: 465–472.
Tong CK, Fuentealba LC, Shah JK, Lindquist RA, Ihrie RA, Guinto CD, et al. A dorsal SHH-dependent domain in the V-SVZ produces large numbers of oligodendroglial lineage cells in the postnatal brain. Stem Cell Reports 2015, 5: 461–470.
Ponti G, Obernier K, Guinto C, Jose L, Bonfanti L, Alvarez-Buylla A. Cell cycle and lineage progression of neural progenitors in the ventricular-subventricular zones of adult mice. Proc Natl Acad Sci U S A 2013, 110: E1045–E1054.
Balordi F, Fishell G. Hedgehog signaling in the subventricular zone is required for both the maintenance of stem cells and the migration of newborn neurons. J Neurosci 2007, 27: 5936–5947.
Balordi F, Fishell G. Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. J Neurosci 2007, 27: 14248–14259.
Yabut OR, Fernandez G, Huynh T, Yoon K, Pleasure SJ. Suppressor of fused is critical for maintenance of neuronal progenitor identity during corticogenesis. Cell Rep 2015, 12: 2021–2034.
Zhang L, Mubarak T, Chen Y, Lee T, Pollock A, Sun T. Counter-balance between Gli3 and miR-7 is required for proper morphogenesis and size control of the mouse brain. Front Cell Neurosci 2018, 12: 259.
Shao W, Yang J, He M, Yu XY, Lee CH, Yang Z, et al. Centrosome anchoring regulates progenitor properties and cortical formation. Nature 2020, 580: 106–112.
Roy A, Murphy RM, Deng M, MacDonald JW, Bammler TK, Aldinger KA, et al. PI3K-Yap activity drives cortical gyrification and hydrocephalus in mice. Elife 2019, 8: e45961.
Roy A, Skibo J, Kalume F, Ni J, Rankin S, Lu Y, et al. Mouse models of human PIK3CA-related brain overgrowth have acutely treatable epilepsy. Elife 2015, 4: e12703.
Marín O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci 2012, 13: 107–120.
Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 2005, 6: 312–324.
Rubenstein JLR, Merzenich MM. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2003, 2: 255–267.
Marín O. Parvalbumin interneuron deficits in schizophrenia. Eur Neuropsychopharmacol 2024, 82: 44–52.
Mack NR, Deng S, Yang SS, Shu Y, Gao WJ. Prefrontal cortical control of anxiety: Recent advances. Neuroscientist 2023, 29: 488–505.
Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry 2019, 24: 1248–1257.
Dienel SJ, Lewis DA. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol Dis 2019, 131: 104208.
DeFelipe J, López-Cruz PL, Benavides-Piccione R, Bielza C, Larrañaga P, Anderson S, et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci 2013, 14: 202–216.
Fishell G, Kepecs A. Interneuron types as attractors and controllers. Annu Rev Neurosci 2020, 43: 1–30.
Gelman DM, Marín O. Generation of interneuron diversity in the mouse cerebral cortex. Eur J Neurosci 2010, 31: 2136–2141.
Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci 2006, 7: 687–696.
Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 2011, 71: 45–61.
Hu JS, Vogt D, Sandberg M, Rubenstein JL. Cortical interneuron development: A tale of time and space. Development 2017, 144: 3867–3878.
Miyoshi G, Hjerling-Leffler J, Karayannis T, Sousa VH, Butt SJB, Battiste J, et al. Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J Neurosci 2010, 30: 1582–1594.
Batista-Brito R, Fishell G. The developmental integration of cortical interneurons into a functional network. Curr Top Dev Biol 2009, 87: 81–118.
Fishell G, Rudy B. Mechanisms of inhibition within the telencephalon: “where the wild things are.” Annu Rev Neurosci 2011, 34: 535–567.
Lim L, Mi D, Llorca A, Marín O. Development and functional diversification of cortical interneurons. Neuron 2018, 100: 294–313.
Nord AS, Pattabiraman K, Visel A, Rubenstein JLR. Genomic perspectives of transcriptional regulation in forebrain development. Neuron 2015, 85: 27–47.
Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: Dependence on Dlx genes. Science 1997, 278: 474–476.
Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: Evidence for a transformation of the pallidum into the striatum. Development 1999, 126: 3359–3370.
Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci 1999, 19: 7881–7888.
Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development 2001, 128: 3759–3771.
Liu Z, Zhang Z, Lindtner S, Li Z, Xu Z, Wei S, et al. Sp9 regulates medial ganglionic eminence-derived cortical interneuron development. Cereb Cortex 2019, 29: 2653–2667.
Wei S, Du H, Li Z, Tao G, Xu Z, Song X, et al. Transcription factors Sp8 and Sp9 regulate the development of caudal ganglionic eminence-derived cortical interneurons. J Comp Neurol 2019, 527: 2860–2874.
Zhang Q, Zhang Y, Wang C, Xu Z, Liang Q, An L, et al. The zinc finger transcription factor Sp9 is required for the development of striatopallidal projection neurons. Cell Rep 2016, 16: 1431–1444.
Xu Z, Liang Q, Song X, Zhang Z, Lindtner S, Li Z, et al. SP8 and SP9 coordinately promote D2-type medium spiny neuron production by activating Six3 expression. Development 2018, 145: dev165456.
Ma T, Zhang Q, Cai Y, You Y, Rubenstein JLR, Yang Z. A subpopulation of dorsal lateral/caudal ganglionic eminence-derived neocortical interneurons expresses the transcription factor Sp8. Cereb Cortex 2012, 22: 2120–2130.
Long JE, Swan C, Liang WS, Cobos I, Potter GB, Rubenstein JLR. Dlx1&2 and Mash1 transcription factors control striatal patterning and differentiation through parallel and overlapping pathways. J Comp Neurol 2009, 512: 556–572.
Flames N, Pla R, Gelman DM, Rubenstein JLR, Puelles L, Marín O. Delineation of multiple subpallial progenitor domains by the combinatorial expression of transcriptional codes. J Neurosci 2007, 27: 9682–9695.
Li J, Wang C, Zhang Z, Wen Y, An L, Liang Q, et al. Transcription factors Sp8 and Sp9 coordinately regulate olfactory bulb interneuron development. Cereb Cortex 2018, 28: 3278–3294.
Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: Implications for striatal and olfactory bulb neurogenesis. J Neurosci 2003, 23: 167–174.
Shang Z, Yang L, Wang Z, Tian Y, Gao Y, Su Z, et al. The transcription factor Zfp503 promotes the D1 MSN identity and represses the D2 MSN identity. Front Cell Dev Biol 2022, 10: 948331.
Li Z, Shang Z, Sun M, Jiang X, Tian Y, Yang L, et al. Transcription factor Sp9 is a negative regulator of D1-type MSN development. Cell Death Discov 2022, 8: 301.
Su Z, Wang Z, Lindtner S, Yang L, Shang Z, Tian Y, et al. Dlx1/2-dependent expression of Meis2 promotes neuronal fate determination in the mammalian striatum. Development 2022, 149: dev200035.
Song X, Chen H, Shang Z, Du H, Li Z, Wen Y, et al. Homeobox gene Six3 is required for the differentiation of D2-type medium spiny neurons. Neurosci Bull 2021, 37: 985–998.
Yang L, Su Z, Wang Z, Li Z, Shang Z, Du H, et al. Transcriptional profiling reveals the transcription factor networks regulating the survival of striatal neurons. Cell Death Dis 2021, 12: 262.
Zhang Z, Wei S, Du H, Su Z, Wen Y, Shang Z, et al. Zfhx3 is required for the differentiation of late born D1-type medium spiny neurons. Exp Neurol 2019, 322: 113055.
Wen Y, Zhang Z, Li Z, Liu G, Tao G, Song X, et al. The PROK2/PROKR2 signaling pathway is required for the migration of most olfactory bulb interneurons. J Comp Neurol 2019, 527: 2931–2947.
Cai Y, Zhang Q, Wang C, Zhang Y, Ma T, Zhou X, et al. Nuclear receptor COUP-TFII-expressing neocortical interneurons are derived from the medial and lateral/caudal ganglionic eminence and define specific subsets of mature interneurons. J Comp Neurol 2013, 521: 479–497.
Bayer SA, Altman J (2023) The Human Brain during the Second Trimester 190– to 210–mm Crown-Rump Lengths. CRC Press, Boca Raton.
Letinic K, Zoncu R, Rakic P. Origin of GABAergic neurons in the human neocortex. Nature 2002, 417: 645–649.
Petanjek Z, Berger B, Esclapez M. Origins of cortical GABAergic neurons in the cynomolgus monkey. Cereb Cortex 2009, 19: 249–262.
Jakovcevski I, Mayer N, Zecevic N. Multiple origins of human neocortical interneurons are supported by distinct expression of transcription factors. Cereb Cortex 2011, 21: 1771–1782.
Yu X, Zecevic N. Dorsal radial glial cells have the potential to generate cortical interneurons in human but not in mouse brain. J Neurosci 2011, 31: 2413–2420.
Zecevic N, Hu F, Jakovcevski I. Interneurons in the developing human neocortex. Dev Neurobiol 2011, 71: 18–33.
Hansen DV, Lui JH, Flandin P, Yoshikawa K, Rubenstein JL, Alvarez-Buylla A, et al. Non-epithelial stem cells and cortical interneuron production in the human ganglionic eminences. Nat Neurosci 2013, 16: 1576–1587.
Molnár Z, Butt SJB. Best-laid schemes for interneuron origin of mice and men. Nat Neurosci 2013, 16: 1512–1514.
Laclef C, Métin C. Conserved rules in embryonic development of cortical interneurons. Semin Cell Dev Biol 2018, 76: 86–100.
Shi Y, Wang M, Mi D, Lu T, Wang B, Dong H, et al. Mouse and human share conserved transcriptional programs for interneuron development. Science 2021, 374: eabj6641.
Yu Y, Zeng Z, Xie D, Chen R, Sha Y, Huang S, et al. Interneuron origin and molecular diversity in the human fetal brain. Nat Neurosci 2021, 24: 1745–1756.
Delgado RN, Allen DE, Keefe MG, Mancia Leon WR, Ziffra RS, Crouch EE, et al. Individual human cortical progenitors can produce excitatory and inhibitory neurons. Nature 2022, 601: 397–403.
Kim SN, Viswanadham VV, Doan RN, Dou Y, Bizzotto S, Khoshkhoo S, et al. 2023 Cell lineage analysis with somatic mutations reveals late divergence of neuronal cell types and cortical areas in human cerebral cortex. bioRxiv: 11.06.565899.
Allen DE, Donohue KC, Cadwell CR, Shin D, Keefe MG, Sohal VS, et al. Fate mapping of neural stem cell niches reveals distinct origins of human cortical astrocytes. Science 2022, 376: 1441–1446.
Andrews MG, Siebert C, Wang L, White ML, Ross J, Morales R, et al. LIF signaling regulates outer radial glial to interneuron fate during human cortical development. Cell Stem Cell 2023, 30: 1382-1391.e5.
Wang L, Wang C, Moriano JA, Chen S, Zhang S, Mukhtar T, et al. Molecular and cellular dynamics of the developing human neocortex at single-cell resolution. bioRxiv 2024, https://doi.org/10.1101/2024.01.16.575956.
Chung C, Yang X, Hevner RF, Kennedy K, Vong KI, Liu Y, et al. Cell-type-resolved mosaicism reveals clonal dynamics of the human forebrain. Nature 2024, 629: 384–392.
Pebworth MP, Ross J, Andrews M, Bhaduri A, Kriegstein AR. Human intermediate progenitor diversity during cortical development. Proc Natl Acad Sci U S A 2021, 118: e2019415118.
Rash BG, Duque A, Morozov YM, Arellano JI, Micali N, Rakic P. Gliogenesis in the outer subventricular zone promotes enlargement and gyrification of the primate cerebrum. Proc Natl Acad Sci U S A 2019, 116: 7089–7094.
Fuccillo M, Rallu M, McMahon AP, Fishell G. Temporal requirement for hedgehog signaling in ventral telencephalic patterning. Development 2004, 131: 5031–5040.
Rallu M, Machold R, Gaiano N, Corbin JG, McMahon AP, Fishell G. Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development 2002, 129: 4963–4974.
Ihrie RA, Shah JK, Harwell CC, Levine JH, Guinto CD, Lezameta M, et al. Persistent sonic hedgehog signaling in adult brain determines neural stem cell positional identity. Neuron 2011, 71: 250–262.
Rubenstein JL, Nord AS, Ekker M. DLX genes and proteins in mammalian forebrain development. Development 2024, 151: dev202684.
Lindtner S, Catta-Preta R, Tian H, Su-Feher L, Price JD, Dickel DE, et al. Genomic resolution of DLX-orchestrated transcriptional circuits driving development of forebrain GABAergic neurons. Cell Rep 2019, 28: 2048-2063.e8.
Di Bella DJ, Habibi E, Stickels RR, Scalia G, Brown J, Yadollahpour P, et al. Molecular logic of cellular diversification in the mouse cerebral cortex. Nature 2021, 595: 554–559.
Waclaw RR, Wang B, Pei Z, Ehrman LA, Campbell K. Distinct temporal requirements for the homeobox gene Gsx2 in specifying striatal and olfactory bulb neuronal fates. Neuron 2009, 63: 451–465.
Waclaw RR, Allen ZJ 2nd, Bell SM, Erdélyi F, Szabó G, Potter SS, et al. The zinc finger transcription factor Sp8 regulates the generation and diversity of olfactory bulb interneurons. Neuron 2006, 49: 503–516.
Florio M, Borrell V, Huttner WB. Human-specific genomic signatures of neocortical expansion. Curr Opin Neurobiol 2017, 42: 33–44.
Heide M, Huttner WB. Human-specific genes, cortical progenitor cells, and microcephaly. Cells 2021, 10: 1209.
Pinson A, Huttner WB. Neocortex expansion in development and evolution-from genes to progenitor cell biology. Curr Opin Cell Biol 2021, 73: 9–18.
Florio M, Albert M, Taverna E, Namba T, Brandl H, Lewitus E, et al. Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 2015, 347: 1465–1470.
Heide M, Haffner C, Murayama A, Kurotaki Y, Shinohara H, Okano H, et al. Human-specific ARHGAP11B increases size and folding of primate neocortex in the fetal marmoset. Science 2020, 369: 546–550.
Judson HF. The Eighth Day of Creation: Makers of the Revolution in Biology. New York: Cold Spring Harbor Laboratory Press, 1996.
Jacob F. Evolution and tinkering. Science 1977, 196: 1161–1166.
Bitar M, Kuiper S, O’Brien EA, Barry G. Genes with human-specific features are primarily involved with brain, immune and metabolic evolution. BMC Bioinformatics 2019, 20: 406.
Acknowledgements
I thank Professor John L. Rubenstein for his comments, suggestions, and edits. I am grateful to Lin Yang, Zhejun Xu, Mengge Sun, Yanjing Gao, and Zhuangzhi Zhang for their help in preparing the figures. This review was supported by the Ministry of Science and Technology of China (STI2030-2021ZD0202300); the National Natural Science Foundation of China (32070971, 32100768, 32200776, and 32200792); the Shanghai Municipal Science and Technology Major Project (2018SHZDZX01); ZJ Lab; and the Shanghai Center for Brain Science and Brain-Inspired Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author claims that there are no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Z. The Principle of Cortical Development and Evolution. Neurosci. Bull. (2024). https://doi.org/10.1007/s12264-024-01259-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12264-024-01259-2