
Abstract
Microglia-mediated neuroinflammation is widely perceived as a contributor to numerous neurological diseases and mental disorders including depression. Discs large homolog 1 (Dlg1), an adaptor protein, regulates cell polarization and the function of K+ channels, which are reported to regulate the activation of microglia. However, little is known about the role of Dlg1 in microglia and the maintenance of central nervous system homeostasis. In this study, we found that Dlg1 knockdown suppressed lipopolysaccharide (LPS)-induced inflammation by down-regulating the activation of nuclear factor-κB signaling and the mitogen-activated protein kinase pathway in microglia. Moreover, using an inducible Dlg1 microglia-specific knockout (Dlg1flox/flox; CX3CR1CreER) mouse line, we found that microglial Dlg1 knockout reduced the activation of microglia and alleviated the LPS-induced depression-like behavior. In summary, our results demonstrated that Dlg1 plays a critical role in microglial activation and thus provides a potential therapeutic target for the clinical treatment of depression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is a severe psychiatric condition that affects nearly 15% of people worldwide [1], and it significantly increases the risk of suicide [2]. However, the pathophysiology of MDD is yet to be completely elucidated. Neuroinflammation is increasingly being accepted as a risk factor for emotional disorders, especially depression [3, 4]. Several clinical and preclinical studies have revealed that signatures of neuroinflammation, including activated microglia and inflammatory mediators, are commonly found in patients with depression [4, 5].
Microglia, the resident immune cells of the central nervous system (CNS), play indispensable roles in both health and disease [6]. Upon neuroinflammation, microglia are the first activated cells in the brain parenchyma [7]. They not only participate in the processes of neurodegenerative diseases [8,9,10] but are also involved in psychiatric disorders [11, 12]. Evidence has shown that microglial status is strongly associated with depression. Endotoxin stimulation can activate microglia and thus cause negative affective disorders [13], and microglia-derived pro-inflammatory cytokines are correlated with depression-like behaviors [14]. Furthermore, by targeting serotonin (5-HT), antidepressants inhibit depression-induced microglial activation and cytokine production, and alleviate depression-like symptoms [15,16,17]. Given that microglia-induced neuroinflammation is an important contributor to MDD, a major goal is to understand the precise underlying mechanism of microglial function in MDD.
Discs large homolog 1 (Dlg1, also known as SAP-97) is a member of the membrane-associated guanylate kinase family [18]. It is an adaptor protein that regulates cell polarity, neuronal Kv1 channel localization, and Kv1.3 channel function [19, 20]. Moreover, the Kv1.3 potassium channel has been reported to regulate the function of microglia, and selectively blocking Kv1.3 channels inhibits microglial activation [21]. In the CNS, Dlg1 interacts with AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and NMDA (N-methyl-D-aspartic acid) receptors when they are sorted and transported from the soma to synapses [22]. In the peripheral immune system, Dlg1 is involved in the activation of dendritic cells and T cells, and knockout of Dlg1 inhibits the release of inflammatory cytokines [23, 24]. In addition, Dlg1 contributes to neuropsychiatric disorders, and is regarded as a risk factor for schizophrenia [25]. Despite the multiple roles of Dlg1, its roles in microglial activation and in MDD remain to be clarified.
In this study, we demonstrated that Dlg1 regulates microglial activation by targeting nuclear factor-κB (NF-κB) signaling and the mitogen-activated protein kinase (MAPK) pathway. In vivo, conditional knockout of microglial Dlg1 inhibited microglial activation, decreased inflammatory cytokine levels, and alleviated depression-like behaviors in mice, providing a potential therapeutic strategy to treat or slow down the progression of depression.
Materials and Methods
Mice
CX3CR1CreER mice were purchased from the Jackson Laboratory (Bar Harbor, USA), and Dlg1flox/flox mice were gifted by Dr. Wanli Liu (Tsinghua University, China). The generation of the Dlg1flox/flox mouse has been described [26]. To generate microglia-specific knockout mice, homozygous Dlg1flox/flox mice were crossed with mice expressing tamoxifen (TAM)-inducible Cre-recombinase under the control of the CX3CR1 promoter (CX3CR1CreER mice [27, 28]). Mice were given TAM by intragastric administration at the age of 6 weeks to induce microglia-specific knockout. All mice were housed in a specific pathogen-free environment at the Animal Care Facility in our institute. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Beijing Institute of Basic Medical Sciences.
Cell Culture and Transfection
All procedures were performed as previously described [29]. Briefly, BV2 and HEK293T cell lines were obtained from the American Type Culture Collection (Manassas, USA) and maintained in Dulbecco's modified Eagle's medium (Invitrogen, Waltham, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, USA) and 1% penicillin-streptomycin (Invitrogen). All cells were maintained in a 5% CO2 atmosphere at 37°C.
For stable knockdown of Dlg1 in BV2 cells, shRNAs against Dlg1 (targeting sequence: #A: GTTGCATCATCTGTACTATTC, #B: GCAACCTCTTTCAGGCTTTAA) were used. Briefly, the shRNA was annealed and ligated into the pLKO.1 lentiviral vector (Addgene, Cambridge, USA) and then co-transfected with the viral packaging plasmids VSVG and ΔR812 into HEK293T cells. The viral supernatant was collected, centrifuged, filtered through a 0.45-μm filter, and then used to infected BV2 cells with polybrene (#sc-134220, Santa Cruz Biotechnology, Dallas, USA, 1:1000). Three days after infection, the cells were selected with puromycin.
Plasmid Constructs and Selection of Cells with Stable Expression
Full-length Dlg1 was amplified from a mouse cDNA library by PCR and inserted into the pCDH-CMV-MCS-EF1-Puro expression vector (Sigma-Aldrich, St Louis, USA) using the EcoRI and NotI restriction sites. To generate cells that stably overexpressed Dlg1, BV2 cells were infected with the retroviral vector pCDH encoding Dlg1 and selected with puromycin.
Reverse Transcription and Real-Time PCR
Total RNA was extracted from cultured cells, brain tissue, or sorted cells using TRIzolTM reagent (#15596018, Invitrogen). cDNA was obtained using a one-step first strand cDNA synthesis kit (AE311-03, TransGen Biotech, Beijing, China). The primer sequences used were as follows:
-
GAPDH forward: 5′-AGGTCGGTGTGAACGGATTTG-3′;
-
GAPDH reverse: 5′-TGTAGACCATGTAGTTGAGGTCA-3′;
-
Dlg1 forward: 5′-AGTGACGAAGTCGGAGTGATT-3′;
-
Dlg1 reverse: 5′-GTCAGGGATCTCCCCTTTATCT-3′;
-
TNF-α forward: 5′-CCCTCACACTCAGATCATCTTCT-3′;
-
TNF-α reverse: 5′-GCTACGACGTGGGCTACAG-3′;
-
Il-6 forward: 5′-CCAAGAGGTGAGTGCCTTCCC-3′;
-
Il-6 reverse: 5′-CTGTTGTTCAGACTCTCTCCCT-3′;
-
Il-1β forward: 5′-GCAACTGTTCCTGAACTCAACT-3′;
-
Il-1β reverse: 5′-ATCTTTTGGGGTCCGTCAACT-3′.
Drugs and Treatment
Lipopolysaccharide (LPS; Sigma-Aldrich) was dissolved in saline (0.9% NaCl), and injected intraperitoneally at 1 mg/kg. Tamoxifen (#S1238, Selleckchem, Houston, USA) was dissolved in corn oil. Mice at the age of 6 weeks were given a total dose of 20 mg tamoxifen intragastrically for three consecutive days to induce microglial Dlg1-specific knockout.
Western Blotting
Whole cells and hippocampal tissue were lysed on ice using cell lysis buffer [50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1 mmol/L EDTA, 1% Triton X-100, 0.1% deoxycholate, 0.05% sodium dodecyl sulfate (SDS), and protease inhibitor cocktail]. The lysates were centrifuged at 15,000× g for 15 min at 4°C, then supernatants were mixed with 6× SDS loading buffer and boiled for 10 min. Samples were separated by SDS-PAGE and transferred to a nitrocellulose (NC) membrane which was blocked in 5% milk and subsequently incubated overnight with the primary antibody. Relevant horseradish peroxidase (HRP)-conjugated secondary antibodies were incubated, and an electrochemiluminescence (ECL) detection reagent was applied to the NC membrane. The protein signal was detected using an X-Ray film processor (Optimax, New York, USA). The antibodies used for immunoblotting were as follows: anti-SAP97 (K64/15) (#75-030, Antibodies Incorporated, Davis, USA), anti-iNOS/NOS Type II (#610332, BD Biosciences, San Jose, USA), anti-Phospho-IKKα/β (S176/180, 16A6) (#2697P), anti-p38 MAPK (#9212), anti-phospho-p38 MAPK (Thr180/Tyr182) (#4511), anti-JNK2 (56G8) (#9258), anti-phospho-SAPK/JNK (Thr183/Tyr185) (#9251), anti-IκBα (44D4) (#4812), and anti-phospho-IκBα (Ser32) (#2859) (Cell Signaling Technology, Beverly, USA), anti-IKKα (CHUK) (#A2062, ABclonal Technology, Wuhan, China), anti-GAPDH (#CW0266A, CWBiotech, Beijing, China), and anti-β-actin (60008-1-Ig, Proteintech Group, Campbell Park, Chicago, USA).
Dual-Luciferase Reporter System
The NF-κB reporter was generated in our laboratory. Briefly, the NF-κB promoter was cloned into a pGL3-luciferase reporter vector (Promega, Madison, USA). The pCMV-Renilla plasmid and NF-κB reporter were co-transfected into HEK293T cells using Lipofectamine 2000 transfection reagent (#11668019, Invitrogen). Sixteen hours after transfection, the cells were lysed and luciferase activity was measured using a dual-luciferase reporter detection system (Promega).
Immunohistochemistry and Immunofluorescence
After anesthesia (0.7% pentobarbital sodium intraperitoneal injection), each mouse was perfused with normal saline and the brain was removed, fixed in 4% paraformaldehyde for 24 h, and dehydrated overnight in 30% sucrose in phosphate buffered saline (PBS). The whole brain was embedded in optimal cutting temperature compound (OCT) and sectioned on a freezing microtome (CM3050S, Leica, Wetzlar, Germany). The coronal sections were incubated overnight with anti-goat IBA1 antibody (1:500; Cat. 019-19741, WAKO, Japan) and anti-mouse GFAP antibody (1:400; MAB360, Millipore, Darmstadt, Germany). Immunostaining was visualized with DAB kit (Zhongshanjinqiao, Beijing, China) after reaction with hydrogen peroxide. All stained sections were examined under a laser scanning confocal microscope (A1, Nikon, Tokyo, Japan).
Open Field Test (OFT)
The mice were kept in the test room for 2 h in advance for preconditioning. During the experiment, each mouse was placed in a 32 × 32 cm2 test box. The test program was started to record the spontaneous activity of the mouse during 5 min and to count the total distance travelled and the time spent in the central area.
Elevated Plus Maze (EPM)
The mice were kept in the test room for 2 h in advance for preconditioning. During the test, each mouse was gently placed in the center area facing the open arm. The recording software was then started to record the activity time and the number of entries into the open arm during 5 min.
Tail Suspension Test (TST)
The mice were kept in the test room for 2 h in advance for preconditioning. We stuck medical tape to the tail tip, and the mouse was hung on the instrument with a clip. The experimental time was 6 min and the immobility time of each mouse was recorded in the last 4 min of the test.
Forced Swimming Test (FST)
The day before the experiment, the mice were allowed to swim in water for 5 min. On the day of the experiment, the mice were kept in the test room for preconditioning. During the test, each mouse was placed in a beaker (volume 3 L) filled with water at 18–21 °C. The total test time was 6 min. The immobility time was recorded during the last 4 min of the test.
Statistical Analysis
Data are expressed as the mean ± SEM from at least two independent experiments. Statistical differences between the test and control values were analyzed using Student’s t-test. For multiple comparisons, statistical differences were analyzed by applying ordinary one-way ANOVA (Tukey’s multiple comparison test). Data were considered significant as follows: *P < 0.05, **P < 0.01, and ***P < 0.001. Statistical analysis was performed using GraphPad Prism (version 6.07, GraphPad, San Diego, USA).
Results
Knockdown of Dlg1 in Microglia Inhibits LPS-induced Inflammation
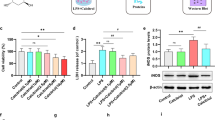
LPS is a commonly-used agonist that induces the activation of microglia. This process is characterized by the overwhelming release of pro-inflammatory cytokines [30, 31]. To investigate the role of Dlg1 in microglial activation, we exposed BV2 cells with Dlg1 knockdown to LPS (1 μg/mL) at different time points (Fig. 1A). Western blot analysis showed that the iNOS protein level was significantly lower in Dlg1-knockdown cells than in control cells (Fig. 1B, C). In addition, knockdown of Dlg1 inhibited the up-regulation of pro-inflammatory cytokines, including tumor necrosis factor (TNF-α), interleukin-1 beta (IL-1β), and IL-6 (Fig. 1D–H). These results suggest that Dlg1 is involved in LPS-induced inflammation, and knockdown of Dlg1 in microglia significantly reduces inflammatory cytokine levels.

Microglial Dlg1 knockdown inhibits LPS-induced inflammation. A Cartoon of LPS stimulation in BV2 cells. B Immunoblots of iNOS, Dlg1, and GAPDH proteins in control and Dlg1-knockdown microglial cells after exposure to LPS (1 µg/mL) for the indicated times. C–F RT-PCR analysis of Dlg1, TNF-α, IL-1β, and IL-6 mRNA levels in control and Dlg1-knockdown microglial cells after exposure to LPS (1 µg/mL) for the indicated times. G, H ELISA analysis of secreted TNF-α and IL-6 levels in control and Dlg1-knockdown BV2 cells after exposure to LPS (1 µg/mL) for the indicated times. I Immunoblots of iNOS, Dlg1, and GAPDH proteins in control and Dlg1-overexpressing microglial cells after exposure to LPS (1 µg/mL) for the indicated times. J–L RT-PCR analysis of TNF-α, IL-1β, and IL-6 mRNA levels in control and Dlg1-overexpressing microglial cells. At least three independent experiments were carried out. M, N ELISA analysis of secreted TNF-α and IL-6 levels in control and Dlg1-overexpressing BV2 cells after exposure to LPS (1 µg/mL) for the indicated times. Data are presented as the mean ± SEM; *P <0.05, **P <0.01, ***P <0.001, two-tailed unpaired Student’s t-test.
To support our findings, we overexpressed Dlg1 in BV2 cells, and these cells were also exposed to LPS at different time points. Consistently, both the iNOS protein level and the cytokine levels were significantly increased when Dlg1 was overexpressed (Fig. 1I–N). Taken together, these results indicate that Dlg1 plays an important role in microglia-mediated inflammation.
Dlg1 Participates in Microglia-mediated Inflammation by Targeting NF-κB Signaling and the MAPK Pathway
It has been well corroborated in previous studies that LPS is an activator of NF-κB signaling and the MAPK pathway [30, 32]. To study the mechanism underlying Dlg1 and microglia-mediated inflammation, we used a short-term LPS challenge, and found that the phosphorylation levels of various pathway proteins declined markedly in Dlg1-knockdown BV2 cells, including P-IKKα/β, P-IκBα, P-JNK, and P-P38 (Fig. 2A). Correspondingly, overexpression of Dlg1 significantly increased the levels of these proteins (Fig. 2B). These results suggest that Dlg1 participates in microglia-mediated inflammation by regulating NF-κB signaling and the MAPK pathway. Since alterations in Dlg1 affect the protein levels of NF-κB signaling, we then asked whether Dlg1 affects its activation. Using a dual-luciferase reporter system, we found that the NF-κB promoter luciferase showed enhanced activity upon overexpression of Dlg1 (Fig. 2C). Collectively, these results indicate that Dlg1 regulates NF-κB signaling and the MAPK pathway and thus plays an important role in microglia-mediated inflammation.

Dlg1 regulates the NF-κB signaling pathway in microglia. A, B Immunoblots of P-IKKα/β, IKKα, P-JNK, JNK2, P-P38, P38, P-IκBα, IκBα, Dlg1, and GAPDH proteins in control, Dlg1-knockdown (A) and Dlg1-overexpressing (OE; B) microglial cells after exposure to LPS (1 µg/mL) for the indicated times. C Quantitative analysis of the effect of Dlg1 overexpression on NF-κB luciferase activity. D Immunoblots of P-IKKα/β, IKKα, P-JNK, JNK2, P-P38, P38, Dlg1, and GAPDH proteins in control and Dlg1-knockdown microglial cells after exposure to LPS (1 µg/mL, 60 min) and KCl (5, 30, 45, and 60 mmol/L). E, F RT-PCR analysis of TNF-α and IL-6 mRNA levels in control and Dlg1-knockdown microglia. Data are presented as the mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed unpaired Student’s t-test.
Previous studies have shown that Dlg1 is an adaptor protein of the Kv1.3 potassium channel by ensuring K+ outflow and subsequent Ca2+ inflow events [22, 23]. To determine whether Dlg1 regulates microglia-mediated inflammation depending on the activity of K+ channels, we applied a serial concentration gradient of K+ culture medium to inhibit K+ channel activity [33] and found that the decreased levels of p-IKKα/β, P-JNK, and P-P38 regulated by Dlg1 knockdown diminished as the concentration of K+ increased (Fig. 2D). Furthermore, the levels of the inflammatory cytokines TNF-α and IL-6 were significantly inhibited when the K+ channel activity was blocked by high concentrations of K+ (Fig. 2E, F). Together, these results indicate that Dlg1 regulates microglia-mediated inflammation in a K+-dependent manner.
Microglial Dlg1 Knockout Alleviates Depression-like Behavior
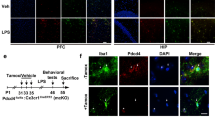
Given that Dlg1 knockdown significantly reduced the inflammatory response in microglia, we next sought to explore the role of Dlg1 in depression. We generated mice with inducible microglia-specific knockout of Dlg1 (Dlg1flox/flox; CX3CR1CreER, abbreviated as cKO mice), and used tamoxifen to induce Cre-recombinase activity in microglia (Fig. 3A). Compared to Dlg1flox/flox mice, the Dlg1 level in cKO mice decreased significantly (Fig. 3B). The LPS-induced depression model has been commonly used in studies based on the causal link between neuroinflammation and depression [34,35,36]. In our experiments, we applied an LPS challenge after tamoxifen administration to induce depression-like behavior in Dlg1flox/flox and cKO mice as assessed by behavioral tests (Fig. 3A). As shown in Fig. 3C–H, treatment with LPS decreased the center time and total distance in the OFT, and decreased the open arm entries and time in the EPM. However, cKO mice did not show significant differences from Dlg1flox/flox mice, suggesting that microglial Dlg1 knockout does not impair LPS-induced anxiety-like behaviors.

Microglial Dlg1 knockout alleviates LPS-induced depression-like behavior in mice. A Timeline of drug administration and behavioral tests. B RT-PCR analysis of microglial Dlg1 mRNA levels from Dlg1flox/flox (n = 3) and cKO (n = 3) mice. C Representative images showing the activity trajectories of mice in the open field test (OFT). D Time spent in the central area by Dlg1flox/flox (n = 11) and cKO (n = 7) mice in the OFT 24 h after saline or LPS (1 mg/kg) injection. E Total distance travelled in the OFT by Dlg1flox/flox (n = 11) and cKO (n = 7) mice. F Representative images showing the activity trajectories of mice in the elevated plus maze (EPM). G, H Numbers of entries into open arms (G) and time spent in the center (H) in the EPM. I Cartoon of the tail suspension test (TST). J Immobility times in the TST. K Cartoon of forced-swimming test (FST). L Immobility times in the FST. Data are presented as the mean ± SEM; *P < 0.05; **P < 0.01, ***P < 0.001, two-tailed unpaired Student’s t-test.
However, in the TST, cKO mice had a shorter immobility time than Dlg1flox/flox mice, suggesting that microglial Dlg1 knockout alleviates the depression-like behavior (Fig. 3I, J). To further characterize the role of Dlg1, we used the FST. Consistent with the results in the TST, microglial Dlg1 knockdown significantly decreased the immobility time, suggesting an improvement in depression-like behavior (Fig. 3K, L). Together, these results demonstrate that deletion of microglial Dlg1 alleviates inflammation-induced depression-like behaviors in mice.
Dlg1 Knockout Inhibits Microglial Activation In Vivo
We next investigated why Dlg1 knockout alleviates depression-like behavior and how microglia change in cKO mice. To address this, we performed IBA1 immunofluorescence staining and found that microglia were robustly activated in the brain of mice injected with LPS, including changes in density and morphology (Fig. 4A, B). Microglial Dlg1 knockout significantly rescued the LPS-induced reduction in branch number and length, as well as increasing the soma area (Fig. 4C–F). Furthermore, LPS injection increased microglial numbers in the hippocampus of Dlg1flox/flox mice, and Dlg1 microglial conditional knockout significantly reduced this increase (Fig. 4G, H), with no effect on astrocyte number (Fig. 4G, I). These results suggest that Dlg1 knockout in microglia markedly inhibits microglial activation in the mouse brain.

Microglial Dlg1 knockout impedes microglial activation in vivo. A Immunofluorescent staining of IBA1 and GFAP in the hippocampal DG area of Dlg1flox/flox and cKO mice with or without LPS exposure (1 mg/kg) (scale bar, 10 μm). B Images and schematics of microglial skeleton analysis. C–F Quantitative analysis of number of microglial branches (C), average branch length (D), total branch length (E), and soma area (F). G Immunohistochemical staining of IBA1 and GFAP in the hippocampus of Dlg1flox/flox and cKO mice (scale bar, 100 μm). H, I Quantitative analysis of IBA1-positive (H) and GFAP-positive cell numbers (I). Data are presented as the mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed unpaired Student’s t-test.
To determine whether inflammatory cytokines also declined, we measured the protein levels of iNOS and IBA1 in the hippocampus and found that upon LPS challenge they were significantly reduced in cKO mice, with a decreasing trend of GFAP levels compared with Dlg1flox/flox mice (Fig. 5A–D). Moreover, we found that the inflammatory cytokines TNF-α and IL-6 were significantly reduced in cKO mice (Fig. 5E, F), with a decreasing trend of IL-1β levels (Fig. 5G).

Microglial Dlg1 knockout decreases inflammation in the mouse brain. A Immunoblots of iNOS, GFAP, IBA1, and GAPDH proteins in the hippocampus of Dlg1flox/flox and cKO mice. B–D Quantitative analysis of iNOS, IBA1, and GFAP protein levels normalized to GAPDH. E–G RT-PCR analysis of TNF-α, IL-1β, and IL-6 mRNA levels in the hippocampus of Dlg1flox/flox and cKO mice. H Model showing the role of Dlg1 in inflammation-induced depression-like behaviors in mice. Data are presented as the mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed unpaired Student’s t-test.
These results collectively indicate that microglial conditional knockout of Dlg1 inhibits microglial activation, and thus alleviates the depression-like behavior induced by neuroinflammation (Fig. 5H).
Discussion
As a representative of bacterial infection, systemic LPS injection (1 mg/kg) causes time-dependent behavioral changes in mice. Pro-inflammatory cytokines in the blood peak at ~2 h, and depression-like behaviors occur 24 h later [14, 37,38,39]. In this study, we used LPS injection to create a depression-like mouse model. Microglial Dlg1 deletion ameliorates the symptoms of depression in mice, and the hippocampus is relatively vulnerable to acute inflammatory attack during depression [34]. Our results demonstrated that Dlg1 knockout reduced microglial activation and inflammatory cytokine levels in the hippocampus, suggesting that Dlg1 functions as an important regulator of inflammation-induced depression.
Neuroinflammation is involved in the regulation of motivational and emotional states in various pathological processes. The increased expression and release of inflammatory mediators, generally manifested as pro-inflammatory cytokines, acute phase proteins, chemokines, and adhesion molecules, are related to human depressive symptoms and negative affective states in mice [40,41,42,43,44,45]. Elevated levels of IL-6, IL-1β and TNF-α are found in both peripheral blood and cerebrospinal fluid [40, 46,47,48,49]. Moreover, the concentrations of these cytokines in plasma are positively correlated with the severity of depressive symptoms [41, 50]. Furthermore, previous studies have revealed that functional allelic variants of the IL-1β and TNF-α genes increase the risk of depression and are associated with decreased responsiveness to antidepressant therapy [51, 52]. Mechanistically, the increased pro-inflammatory cytokines mediated by NF-κB signaling [53] cause long-term alterations in neuropeptide and neurotransmitter synthesis, leading to metabolic changes in serotonin, dopamine, and other related neurotransmitters [54, 55]. Moreover, the MAPK pathway is associated with dysregulation of the hypothalamic–pituitary–adrenal axis, one of the strongest biological correlates of MDD [56, 57]. Here, we found that the NF-κB signaling and MAPK pathways were involved in microglia-mediated inflammation. Knockout of Dlg1 in microglia inhibited the activation of the NF-κB signaling and MAPK pathways, and markedly alleviated depression-like behaviors. Numerous studies have revealed that voltage-gated K+ channels are involved in microglial activation [58, 59]. In this study, we found that the effects of Dlg1 were partially rescued by incubation with high concentrations of K+, suggesting that K+ flux is involved in the effect of Dlg1 in microglia. However, the target and the regulatory mechanism of Dlg1 in microglial activation need to be further investigated.
Microglia are resident immune cells of the CNS. Our recent studies showed that inhibition of microglial activation-induced neuroinflammation alleviates brain injury in multiple neurodegenerative diseases and psychological disorders [9, 10, 60, 61]. Moreover, a recent study showed that microglial activation regulates negative emotions through prostaglandin-dependent striatal neurons, and thus plays a key role in the development of major depression [62]. In this study, we found that knockout of Dlg1 in microglia alleviated neuroinflammation-induced depression-like behaviors in mice, providing a novel way to interfere with microglial activation and an alternative treatment strategy for depression.
Conclusions
In summary, our study demonstrated that knockdown of Dlg1 inhibits the activation of NF-κB signaling and the MAPK pathway in microglia, and consequently reduces microglial activation and the release of pro-inflammatory cytokines. Furthermore, deletion of microglial Dlg1 alleviates inflammation-induced depression-like symptoms in mice. Our findings demonstrate that Dlg1 plays a vital role in microglial activation and provides a potential therapeutic target for the treatment of depression.
References
Andrade L, Caraveo-Anduaga JJ, Berglund P, Bijl RV, de Graaf R, Vollebergh W. The epidemiology of major depressive episodes: Results from the International Consortium of Psychiatric Epidemiology (ICPE) Surveys. Int J Methods Psychiatr Res 2003, 12: 3–21.
Cheng AT, Chen TH, Chen CC, Jenkins R. Psychosocial and psychiatric risk factors for suicide. Case-control psychological autopsy study. Br J Psychiatry 2000, 177: 360–365.
Payne JL, Maguire J. Pathophysiological mechanisms implicated in postpartum depression. Front Neuroendocrinol 2019, 52: 165–180.
Troubat R, Barone P, Leman S, Desmidt T, Cressant A, Atanasova B, et al. Neuroinflammation and depression: A review. Eur J Neurosci 2021, 53: 151–171.
Liang S, Wang Q, Kong X, Deng W, Yang X, Li X, et al. White matter abnormalities in major depression biotypes identified by diffusion tensor imaging. Neurosci Bull 2019, 35: 867–876.
Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med 2017, 23: 1018–1027.
Kaminska B, Mota M, Pizzi M. Signal transduction and epigenetic mechanisms in the control of microglia activation during neuroinflammation. Biochim Biophys Acta 2016, 1862: 339–351.
Zhao SQ, Yin J, Zhou LJ, Yan F, He Q, Huang L, et al. Hippo/MST1 signaling mediates microglial activation following acute cerebral ischemia-reperfusion injury. Brain Behav Immun 2016, 55: 236–248.
Pan RY, Ma J, Kong XX, Wang XF, Li SS, Qi XL, et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-β clearance. Sci Adv 2019, 5: eaau6328. https://doi.org/10.1126/sciadv.aau6328.
Cheng JB, Liao YJ, Xiao L, Wu R, Zhao SQ, Chen H, et al. Autophagy regulates MAVS signaling activation in a phosphorylation-dependent manner in microglia. Cell Death Differ 2017, 24: 276–287.
Rial D, Lemos C, Pinheiro H, Duarte JM, Gonçalves FQ, Real JI, et al. Depression as a glial-based synaptic dysfunction. Front Cell Neurosci 2015, 9: 521.
Liu JF, Wu RY, Li JX. Toll of mental disorders: TLR-mediated function of the innate immune system. Neurosci Bull 2019, 35: 771–774.
Reichenberg A, Yirmiya R, Schuld A, Kraus T, Haack M, Morag A, et al. Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiatry 2001, 58: 445–452.
Zhao XN, Cao FR, Liu Q, Li XS, Xu GY, Liu G, et al. Behavioral, inflammatory and neurochemical disturbances in LPS and UCMS-induced mouse models of depression. Behav Brain Res 2019, 364: 494–502.
Li MM, Li CL, Yu HJ, Cai XX, Shen XB, Sun X, et al. Lentivirus-mediated interleukin-1β (IL-1β) knock-down in the hippocampus alleviates lipopolysaccharide (LPS)-induced memory deficits and anxiety- and depression-like behaviors in mice. J Neuroinflammation 2017, 14: 190.
Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Behav Immun 2012, 26: 469–479.
Li Y, Liu J, Liu X, Su CJ, Zhang QL, Wang ZH, et al. Correction to: Antidepressant-like action of single facial injection of botulinum neurotoxin A is associated with augmented 5-HT levels and BDNF/ERK/CREB pathways in mouse brain. Neurosci Bull 2019, 35: 779–780.
Oliva C, Escobedo P, Astorga C, Molina C, Sierralta J. Role of the MAGUK protein family in synapse formation and function. Dev Neurobiol 2012, 72: 57–72.
Szilágyi O, Boratkó A, Panyi G, Hajdu P. The role of PSD-95 in the rearrangement of Kv1.3 channels to the immunological synapse. Pflugers Arch 2013, 465: 1341–1353.
Tejedor FJ, Bokhari A, Rogero O, Gorczyca M, Zhang J, Kim E, et al. Essential role for dlg in synaptic clustering of Shaker K+ channels in vivo. J Neurosci 1997, 17: 152–159.
Rangaraju S, Raza SA, Pennati A, Deng QD, Dammer EB, Duong D, et al. A systems pharmacology-based approach to identify novel Kv1.3 channel-dependent mechanisms in microglial activation. J Neuroinflammation 2017, 14: 128.
Lin EI, Jeyifous O, Green WN. CASK regulates SAP97 conformation and its interactions with AMPA and NMDA receptors. J Neurosci 2013, 33: 12067–12076.
Dong XJ, Wei LS, Guo XH, Yang ZY, Wu C, Li PY, et al. Dlg1 maintains dendritic cell function by securing voltage-gated K+ channel integrity. J Immunol 2019, 202: 3187–3197.
Zanin-Zhorov A, Lin JQ, Scher J, Kumari S, Blair D, Hippen KL, et al. Scaffold protein Disc large homolog 1 is required for T-cell receptor-induced activation of regulatory T-cell function. Proc Natl Acad Sci U S A 2012, 109: 1625–1630.
Gupta P, Uner OE, Nayak S, Grant GR, Kalb RG. SAP97 regulates behavior and expression of schizophrenia risk enriched gene sets in mouse hippocampus. PLoS One 2018, 13: e0200477. https://doi.org/10.1371/journal.pone.0200477.
Zhou WG, Zhang L, Guoxiang GX, Mojsilovic-Petrovic J, Takamaya K, Sattler R, et al. GluR1 controls dendrite growth through its binding partner, SAP97. J Neurosci 2008, 28: 10220–10233.
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38: 79–91.
Xu ZX, Kim GH, Tan JW, Riso AE, Sun Y, Xu EY, et al. Elevated protein synthesis in microglia causes autism-like synaptic and behavioral aberrations. Nat Commun 2020, 11: 1797.
Li XH, Liao YJ, Dong Y, Li SS, Wang FC, Wu R, et al. Mib2 deficiency inhibits microglial activation and alleviates ischemia-induced brain injury. Aging Dis 2020, 11: 523–535.
Bode JG, Ehlting C, Häussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal 2012, 24: 1185–1194.
Zusso M, Lunardi V, Franceschini D, Pagetta A, Lo R, Stifani S, et al. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J Neuroinflammation 2019, 16: 148.
Sen R, Smale ST. Selectivity of the NF-{kappa}B response. Cold Spring Harb Perspect Biol 2010, 2: a000257.
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38: 1142–1153.
Fang K, Li HR, Chen XX, Gao XR, Huang LL, Du AQ, et al. Corrigendum: quercetin alleviates LPS-induced depression-like behavior in rats via regulating BDNF-related imbalance of copine 6 and TREM1/2 in the hippocampus and PFC. Front Pharmacol 2020, 11: 518.
Li WF, Ali T, Zheng CY, Liu ZZ, He KW, Shah FA, et al. Fluoxetine regulates eEF2 activity (phosphorylation) via HDAC1 inhibitory mechanism in an LPS-induced mouse model of depression. J Neuroinflammation 2021, 18: 38.
Florensa-Zanuy E, Garro-Martínez E, Adell A, Castro E, Díaz Á, Pazos Á, et al. Cannabidiol antidepressant-like effect in the lipopolysaccharide model in mice: Modulation of inflammatory pathways. Biochem Pharmacol 2021, 185: 114433.
O’Connor JC, Lawson MA, André C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2, 3-dioxygenase activation in mice. Mol Psychiatry 2009, 14: 511–522.
Custódio CS, Mello BS, Cordeiro RC, de Araújo FY, Chaves JH, Vasconcelos SM, et al. Time course of the effects of lipopolysaccharide on prepulse inhibition and brain nitrite content in mice. Eur J Pharmacol 2013, 713: 31–38.
Wang Z, Zhang QR, Yuan L, Wang SL, Liu LW, Yang XD, et al. The effects of curcumin on depressive-like behavior in mice after lipopolysaccharide administration. Behav Brain Res 2014, 274: 282–290.
Alesci S, Martinez PE, Kelkar S, Ilias I, Ronsaville DS, Listwak SJ, et al. Major depression is associated with significant diurnal elevations in plasma interleukin-6 levels, a shift of its circadian rhythm, and loss of physiological complexity in its secretion: Clinical implications. J Clin Endocrinol Metab 2005, 90: 2522–2530.
Miller GE, Stetler CA, Carney RM, Freedland KE, Banks WA. Clinical depression and inflammatory risk markers for coronary heart disease. Am J Cardiol 2002, 90: 1279–1283.
Musselman DL, Miller AH, Porter MR, Manatunga A, Gao F, Penna S, et al. Higher than normal plasma interleukin-6 concentrations in cancer patients with depression: Preliminary findings. Am J Psychiatry 2001, 158: 1252–1257.
Kahl KG, Rudolf S, Stoeckelhuber BM, Dibbelt L, Gehl HB, Markhof K, et al. Bone mineral density, markers of bone turnover, and cytokines in young women with borderline personality disorder with and without comorbid major depressive disorder. Am J Psychiatry 2005, 162: 168–174.
Engler H, Brendt P, Wischermann J, Wegner A, Röhling R, Schoemberg T, et al. Selective increase of cerebrospinal fluid IL-6 during experimental systemic inflammation in humans: Association with depressive symptoms. Mol Psychiatry 2017, 22: 1448–1454.
Dantzer R. Neuroimmune interactions: From the brain to the immune system and vice versa. Physiol Rev 2018, 98: 477–504.
Słuzewska A, Rybakowski JK, Laciak M, Mackiewicz A, Sobieska M, Wiktorowicz K. Interleukin-6 serum levels in depressed patients before and after treatment with fluoxetine. Ann N Y Acad Sci. 1995, 762: 474–476.
Tuglu C, Kara SH, Caliyurt O, Vardar E, Abay E. Increased serum tumor necrosis factor-alpha levels and treatment response in major depressive disorder. Psychopharmacology (Berl) 2003, 170: 429–433.
Owen BM, Eccleston D, Ferrier IN, Young AH. Raised levels of plasma interleukin-1beta in major and postviral depression. Acta Psychiatr Scand 2001, 103: 226–228.
Hestad KA, Tønseth S, Støen CD, Ueland T, Aukrust P. Raised plasma levels of tumor necrosis factor alpha in patients with depression: Normalization during electroconvulsive therapy. J ECT 2003, 19: 183–188.
Thomas AJ, Davis S, Morris C, Jackson E, Harrison R, O’Brien JT. Increase in interleukin-1beta in late-life depression. Am J Psychiatry 2005, 162: 175–177.
Yu YW, Chen TJ, Hong CJ, Chen HM, Tsai SJ. Association study of the interleukin-1 beta (C-511T) genetic polymorphism with major depressive disorder, associated symptomatology, and antidepressant response. Neuropsychopharmacology 2003, 28: 1182–1185.
Jun TY, Pae CU, Hoon-Han Chae JH, Bahk WM, Kim KS, et al. Possible association between-G308A tumour necrosis factor-alpha gene polymorphism and major depressive disorder in the Korean population. Psychiatr Genet 2003, 13: 179–181.
Raison CL, Capuron L, Miller AH. Cytokines sing the blues: Inflammation and the pathogenesis of depression. Trends Immunol 2006, 27: 24–31.
Gao HM, Jiang J, Wilson B, Zhang WQ, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: Relevance to Parkinson’s disease. J Neurochem 2002, 81: 1285–1297.
Dunn AJ, Wang J, Ando T. Effects of cytokines on cerebral neurotransmission. Comparison with the effects of stress. Adv Exp Med Biol 1999, 461: 117–127.
McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: Interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev 1999, 20: 435–459.
Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD. p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J Biol Chem 2005, 280: 15649–15658.
Nguyen HM, Grössinger EM, Horiuchi M, Davis KW, Jin LW, Maezawa I, et al. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017, 65: 106–121.
Nguyen HM, di Lucente J, Chen YJ, Cui Y, Ibrahim RH, Pennington MW, et al. Biophysical basis for Kv1.3 regulation of membrane potential changes induced by P2X4-mediated calcium entry in microglia. Glia 2020, 68: 2377–2394.
Dong Y, Li SS, Lu YM, Li XH, Liao YJ, Peng ZX, et al. Stress-induced NLRP3 inflammasome activation negatively regulates fear memory in mice. J Neuroinflammation 2020, 17: 205.
Li SS, Liao YJ, Dong Y, Li XH, Li J, Cheng Y, et al. Microglial deletion and inhibition alleviate behavior of post-traumatic stress disorder in mice. J Neuroinflammation 2021, 18: 7.
Klawonn AM, Fritz M, Castany S, Pignatelli M, Canal C, Similä F, et al. Microglial activation elicits a negative affective state through prostaglandin-mediated modulation of striatal neurons. Immunity 2021, 54: 225-234.e6.
Acknowledgements
We sincerely thank Dr. Wanli Liu (Tsinghua University) for providing Dlg1flox/flox mice. This work was supported by the National Natural Science Foundation of China (82071218, 81630026, and 81930029).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, Z., Li, X., Li, J. et al. Dlg1 Knockout Inhibits Microglial Activation and Alleviates Lipopolysaccharide-Induced Depression-Like Behavior in Mice. Neurosci. Bull. 37, 1671–1682 (2021). https://doi.org/10.1007/s12264-021-00765-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-021-00765-x