
Abstract
Trigeminal neuropathic pain (TNP) is a significant health problem but the involved mechanism has not been completely elucidated. Toll-like receptors (TLRs) have recently been demonstrated to be expressed in the dorsal root ganglion and involved in chronic pain. Here, we show that TLR8 was persistently increased in the trigeminal ganglion (TG) neurons in model of TNP induced by partial infraorbital nerve ligation (pIONL). In addition, deletion or knockdown of Tlr8 in the TG attenuated pIONL-induced mechanical allodynia, reduced the activation of ERK and p38-MAPK, and decreased the expression of pro-inflammatory cytokines in the TG. Furthermore, intra-TG injection of the TLR8 agonist VTX-2337 induced pain hypersensitivity. VTX-2337 also increased the intracellular Ca2+ concentration, induced the activation of ERK and p38, and increased the expression of pro-inflammatory cytokines in the TG. These data indicate that TLR8 contributes to the maintenance of TNP through increasing MAPK-mediated neuroinflammation. Targeting TLR8 signaling may be effective for the treatment of TNP.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The sensation of nociceptive stimuli is mediated by primary sensory neurons in the dorsal root ganglion (DRG) and trigeminal ganglion (TG), which transmit noxious information to the brain via the spinal cord and medulla oblongata, respectively. Painful sensation from the orofacial area is relayed in the TG, which has three major branches: the ophthalmic (V1), maxillary (V2), and mandibular nerves (V3). Trigeminal neuropathic pain (TNP) is usually a result of injury or disease of one or more nerve branches (usually V2 and/or V3) and can be triggered by subtle sensory stimuli to the affected side of the face, such as light mechanical touch, brushing teeth, and chewing [1]. TNP is a chronic state and difficult to treat in clinical practice. Understanding the pathophysiology of TNP is essential for pain management.
In recent years, increasing evidence has revealed that neuroinflammation is involved in the pathological process of neuropathic pain including TNP [2,3,4,5]. After peripheral nerve injury, neuroinflammation occurs at different anatomical locations on the pain transmission pathway, including the TG/DRG and medulla oblongata/spinal cord, which facilitates peripheral and central sensitization [6,7,8,9]. Among inflammatory mediators, cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 are well demonstrated to be increased in the peripheral nervous system after nerve injury and enhance neuronal excitability [7, 10,11,12]. Inhibiting these cytokines using neutralizing antibodies or RNA interference inhibits the pain behavior in several neuropathic pain models [7].
Toll-like receptors (TLRs) play a vital role in the innate and adaptive immune responses [13]. After binding with ligands, TLRs initiate and regulate the inflammatory response via the release of cytokines [14, 15]. The family of TLRs has 12 functional members in mice (TLR1-TLR9 and TLR11-TLR13). Unlike other TLRs, TLR3, 7, 8, and 9 are localized in intracellular compartments of the immune system [14, 16]. However, the evidence shows that TLR7 is localized on the membrane of DRG neurons and regulates itch transmission [17]. In addition, a recent report confirmed that TLR7 is expressed in injured DRG neurons and contributes to neuropathic pain via activating the NF-κB signaling pathway [18]. Recently, TLR9 has been found in macrophages in the DRG and contributes to paclitaxel-induced neuropathic pain in mice [19]. The Tlr8 and Tlr7 genes show high homology to each other and are both located on the X chromosome. However, TLR8 is located in the intracellular endoplasmic reticulum (ER), endosomes, and lysosomes of DRG neurons, and plays an important role in the pathogenesis of spinal nerve injury-induced neuropathic pain [20]. How TLR8 is expressed in the TG after infraorbital nerve injury and whether TLR8 is involved in the TNP have not been investigated.
The mitogen-activated protein kinases (MAPKs), which include extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK), have been reported to contribute to neuroinflammation and chronic pain [21]. In the DRG, the activation of MAPKs including ERK and p38 increases the expression of IL-1β and IL-6, and is involved in post-operative and inflammatory pain [22, 23]. Our earlier study showed that a TLR8 agonist activates ERK in DRG neurons and induces pain hypersensitivity [20]. In this study, we used the partial ligation of the infraorbital nerve (pIONL) model to investigate the role of TLR8 in the TG during the pathogenesis of TNP. We found that TLR8 neurons facilitate TPN via increasing intracellular Ca2+, activating ERK and p38, and further increasing the expression of pro-inflammatory cytokines in the TG after infraorbital nerve injury.
Materials and Methods
Animals and Surgery
ICR male mice weighing 26 g–30 g were provided by the Experimental Animal Center at Nantong University. Tlr8−/− mice were developed by Cyagen Co. (Suzhou, China). Tlr7−/− mice were purchased from the Jackson Laboratory (stock number 008380). All mice were housed in standard clear plastic cages under controlled ambient temperature (22 °C–24 °C) with a reversed 12:12 h dark/light cycle, and allowed ad libitum access to water and food. The experimental and surgical procedures were reviewed and approved by the Animal Care and Use Committee of Nantong University. Animal treatments were performed in accordance with the guidelines of the International Association for the Study of Pain.
The pIONL surgery was as described by Zhang et al. [24, 25]. In brief, mice were anesthetized with sodium pentobarbital (40 mg/kg–50 mg/kg, i.p.), and then the oral cavity was opened to locate the tendon of the left masseter muscle on the upper wall of oral cavity in the supine position. In front of the tendon, an incision (1 mm) was made to expose the infraorbital nerve. The TNP model was established by ligating one-half of the infraorbital nerve (ION) with 6-0 silk suture. The mucous membrane of the incision was closed with Tissue Adhesive (Vetbond; 3M, USA). The sham operation comprised an incision on the mucous membrane without damaging the ION.
DNA Extraction and Genotyping
A piece ~2 mm in diameter was cut from the ear each Tl8−/− or Tlr7−/− mouse under brief anesthesia with isoflurane, and then used to extract DNA with the phenol-chloroform method. The primers for genotyping Tlr8−/− mice were as follows: forward: 5′-GCA GTT GAC GAT GGT TGC ATT-3′, and reverse: 5′-TGA CGT GCT TTT GTC TGC TG-3′. A 50 μL reaction volume of PCR amplification buffer was used, including 200 ng DNA, 25 μL 2×Taq PCR MasterMix (Tiangen Biotech), and 1.0 μmol/L TLR8 genotyping primers. Reactions were initially denatured at 95 °C for 5 min, 35 cycles at 95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s, and then final extension at 72 °C for 2 min. The genotyping protocol for Tlr7−/− mice was as described by the Jackson Laboratory. The primers included the common forward primer for olMR8628: 5′-AGG GTA TGC CGC CAA ATC TAA AG-3′, the wild-type reverse primer for olMR8629: 5′-ACC TTT GTG TGC TCC TGG AC-3′, and the mutant reverse primer for olMR8630: 5′-TCA TTC TCA GTA TTG TTT TGC C-3′. For PCR amplification, 200 ng DNA was used in a 50 μL reaction volume containing 25 μL 2×Taq PCR MasterMix (Tiangen Biotech), and 0.5 μmol/L primers for olMR8628, olMR8629, and olMR8630. Reactions were initially denatured at 94 °C for 2 min, 10 cycles at 94 °C for 20 s, 65 °C for 15 s (0.5 °C decrease per cycle), 68 °C for 10 s, and then 28 cycles at 94 °C for 15 s, 60 °C for 15 s, 72 °C for 10 s, and then final extension at 72 °C for 2 min. Amplicons were separated on 1.5% agarose gel, stained with DuRed (Biotium), and photographed with the GelDoc-It Ts Imaging System (UVP, USA).
Drugs and Administration
VTX-2337 was from Active Biochem (Hong Kong, China). Tlr8 siRNA and negative control siRNA (NC siRNA) were designed by RiboBio [20]. RVG-9R peptide was from AnaSpec (USA). For peri-infraorbital nerve injection, the Tlr8 siRNA (2 µg) or NC siRNA was mixed with RVG-9R peptide (molar ratio 1:10) [20, 26]. Intra-TG injection was performed with a 29 G syringe (Becton, Dickinson and Co., USA). After deep anesthesia with isoflurane, the head of the mouse was held in one hand. The tip of the needle was then inserted through the infraorbital foramen, infraorbital canal, and foramen rotundum, and finally was positioned in the TG [27]. Different doses (10, 50, and 100 ng) of VTX-2337 (5 μL) were slowly injected.
Facial Pain Behavioral Test
The behavioral test environment was kept at 22 °C–24 °C and 40%–60% humidity. Before facial behavioral tests, the mice were habituated to the behavioral test cage for 30 min every day for 3 days in the test environment. Von Frey filaments (0.02 g and 0.16 g) were used to stimulate the whisker pad innervated by the ION, and the responses of the mice were recorded. The filaments were applied 3 times on the ipsilateral whisker pad. The mean nocifensive behavior score of 3 measurements was calculated according to the following criteria: score 0, no response; score 1, exploratory behavior - the mouse detected the von Frey filament; score 2, slight withdrawal response - the mouse slowly moved its face back from the stimulation; score 3, quick and intense withdrawal response with lifting of the paw; score 4, wiping the face with the forepaw < 3 times in the stimulated facial area; score 5, wiping the face with the forepaw > 3 times in the stimulated facial area [28, 29].
RNA Collection and Real-Time RT-PCR
Using the manufacture’s protocol of TRIzol reagent (Invitrogen, Carlsbad, CA, USA), the total RNA in the TG was collected. The quality and quantity of RNA was checked on a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA). The cDNA was reverse-transcribed from RNA using a First Strand cDNA Synthesis Kit (Takara, Japan). Real-time PCR was performed in A&B Applied Biosystems apparatus using SYBR Green Master Mix (Vazyme, China). The following primers were used in the PCR reaction: TLR8 forward: 5′-ACC TGA GCC ACA ATG GCA TTT AC-3′, TLR8 reverse: 5′-TTG CCA TCA TTT GCA TTC CAC-3′; TNF-α forward: 5′-GTT CTA TGG CCC AGA CCC TCA C-3′, TNF-α reverse: 5′-GGC ACC ACT AGT TGG TTG TCT TTG-3′; IL-1β forward: 5′-TCC AGG ATG AGG ACA TGA GCA C -3′, IL-1β reverse: 5′-GAA CGT CAC CCA GCA GGT TA-3′; IL-6 forward: 5′-CCA CTT CAC AAG TCG GAG GCT TA-3′, IL-6 reverse: 5′-CCA GTT TGG TAG CAT CCA TCA TTT C-3′; GAPDH forward: 5′-AAA TGG TGA AGG TCG GTG TGA AC-3′, GAPDH reverse: 5′-CAA CAA TCT CCA CTT TGC CAC TG-3′. The conditions of PCR amplification were set at 95 °C for 30 s, and then 40 cycles (5 s for 95 °C and 30 s for 60 °C). The PCR data were analyzed with StopOne Software v2.3. Melting curves were used to assess the specificity of PCR products. Quantification was performed by normalizing the cycle threshold (Ct) values with GAPDH Ct and analyzed with the 2−ΔΔCt method.
Immunofluorescence
After deep anesthesia with the isoflurane, the mice were transcardially perfused with 0.01 mol/L PBS followed by 4% paraformaldehyde in 0.01 mol/L PBS. The TG was dissected carefully, postfixed in fixative overnight, and then left in 30% sucrose in 0.01 mol/L PBS for 2 days. The TG was embedded in OCT solution and then cut into 15-μm sections on a cryostat (Leica, Germany). The sections were processed for immunofluorescence as we described previously [20]. In brief, the sections were blocked by 5% donkey serum in PBS at room temperature for 1 h. Subsequently, the sections were incubated with primary antibodies against TLR8 (rabbit, 1:500, BosterBio), Tuj1 (mouse, 1:2000, R&D Systems), CD68 (rat, 1:1000, AbD Serotec), ATF3 (mouse, 1:500, Santa Cruz), CGRP (mouse, 1:1000, Sigma-Aldrich), and NF200 (mouse, 1:500, Cell Signaling Technology) in a humidified box at 4 °C overnight. Following three rinses with 0.01 mol/L PBS, the sections were further incubated with the secondary antibodies donkey anti-rabbit Cy3 (1:1000, Jackson ImmunoResearch), donkey anti-rat Alexa Fluor 488 (1:1000, Jackson ImmunoResearch), donkey anti-mouse Alexa Fluor 488 (1:1000, Jackson ImmunoResearch), and IB4-FITC (1:200, Sigma-Aldrich). The fluorescence signals were checked and captured under a fluorescence microscope (Nikon Eclipse Ni-E, Japan). The fluorescence images were analyzed with ImageJ (NIH, USA).
Western Blot
The TGs from WT and Tlr8−/− mice were homogenized in lysis buffer containing protease and phosphatase inhibitors (Sigma-Aldrich). The protein concentration was checked using the BCA Protein Assay (Pierce Biotechnology, USA). Thirty micrograms of protein was loaded in each lane of SDS-PAGE gel, and then transferred to polyvinylidene fluoride membrane. After blocking by 5% skim milk, the membrane was incubated with the primary antibody against pERK (rabbit, 1:500, Cell Signaling Technology), ERK (rabbit, 1:500, Cell Signaling Technology), pp38 (rabbit, 1:500, Cell Signaling Technology), or p38 (rabbit, 1:500, Cell Signaling Technology). The membrane was further incubated with IRDye 800CW secondary antibody (goat-anti-rabbit, 1:10000, LI-COR) and the images captured with an Odyssey CLx system (Odyssey, USA). The sizes of bands were evaluated by the pre-stained protein marker (Thermo Fisher Scientific), and the intensity of bands was calculated by ImageJ.
HEK293 Cell Culture and Transfection
HEK293 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (Bio Whittaker Europe, Vervier, Belgium) with 10% (vol/vol) fetal calf serum (PAA, Linz, Austria) and 0.5% penicillin/streptomycin at 37 °C in a humidity-controlled incubator with 5% (vol/vol) CO2. Cells were transfected with the TLR8 or control plasmids using Lipofectamine 2000 (Invitrogen, Netherlands) at 80% confluence. Transfected cells were cultured in the same growth medium for 36 h before Ca2+ measurements.
TG Neuron Culture
After anesthesia using isoflurane, the cranium and brain of mice (4–6 weeks old) were quickly removed, and the TGs rapidly collected [30]. The meninges and connective tissue on the TG were carefully stripped in an ice-cold oxygenated balanced salt solution (BSS, in mmol/L: 125 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 2.4 CaCl2, 1.2 MgCl2, 5 HEPES, pH 7.2 and osmolarity 300 mOsm), and each TG was shredded with scissors. The TG tissue was kept at 37 °C for 90 min in oxygenated aCSF which contained collagenase (3.0 mg/mL, Roche) and dispase-II (2.4 units/mL, Roche), and then washed twice with standard aCSF. TG neurons were separated mechanically with polished glass pipettes, and cultivated on adhesive coverslips (diameter, 13 mm) in 24-well plates. The dissociated TG neurons in each well were incubated in aCSF at 37 °C (humidified 95% O2 and 5% CO2). After 24 h incubation, TG neurons were used for Ca2+ imaging measurements.
Ca2+ Imaging
HEK 293 cells or TG neurons were loaded with Fura-2AM (2 µmol/L, Molecular Probes) mixed with 0.02% pluronic (Life Technologies) for 90 min at room temperature in darkness. Coverslips with HEK 293 cells or TG neurons were washed with BSS and then placed on an inverted fluorescence microscope (Olympus IX73, Japan). The fluorescence emission at 510 nm with excitation at 340 nm and 380 nm was detected at 2-s intervals using a computer-controlled F4500 fluorescence spectrophotometer (Hitachi, Japan). Wavelength selection and the timing of excitation and acquisition of images were controlled using the Metafluor program (Molecular Devices). Digital images were stored on hard disk for off-line analysis. The ratio of fluorescence intensities (λ340/λ380) at these two wavelengths was recorded as the relative level of intracellular Ca2+.
Quantification and Statistics
All results are shown as mean ± SEM. The behavioral data were analyzed by two-way repeated measures (RM) ANOVA followed by the Bonferroni test as the post-hoc multiple comparison analysis. For the analysis of TLR8+ neurons in the TG, immunofluorescent images of TG were captured, and the number of TG neurons was counted using a computer-assisted imaging analysis system (ImageJ). For western blot, the density of specific bands was measured with ImageJ, and the intensity of the background was subtracted in each lane. The levels of pERK and pp38 were normalized to total ERK and p38, respectively. Differences between groups were compared using one-way ANOVA followed by the Bonferroni test or using Student’s t-test if only 2 groups were compared. The criterion for statistical significance was set at P < 0.05.
Results
TLR8 Expression is Increased in TG Neurons After pIONL-Induced TNP
Before and after pIONL, we used the numerical value of the facial nocifensive behavior score, which was induced by a 0.02 g (or 0.16 g) von Frey filament stimulation of the whisker pad, to evaluate the degree of TNP [28]. Behavioral data showed that pIONL increased the nocifensive behavior score stimulated with a 0.02 g von Frey filament (Fig. 1A), starting from day 3 and lasting for > 28 days after the operation (Fig. 1A). The scores for a 0.16 g filament were similar (data not shown). We then checked the expression of TLR8 in the TG after pIONL. Real-time PCR showed that the expression of Tlr8 mRNA was increased at days 3, 10, and 21 after pIONL (Fig.1B). Immunostaining showed a few TLR8+ neurons scattered in the TG of naïve and sham-operated mice, but this was dramatically increased in pIONL mice at day 10 (Fig. 1C–E). Statistical data showed that TLR8+ cells were significantly increased at pIONL day 10 (Fig. 1F). Double immunostaining showed that TLR8 was totally co-localized with the neuronal marker Tuj1 (Fig. 1G), but not with the macrophage marker CD68 (Fig. 1H), suggesting the neuronal expression of TLR8. The cell-size distribution showed that TLR8 was expressed mainly in small-diameter neurons (< 300 µm2, 70.8%), moderately in medium-diameter neurons (300 µm2–600 µm2, 27.8%), and rarely in large-diameter neurons (> 600 µm2, 1.4%, Fig. 1I). Further double staining showed that TLR8 was highly co-localized with the marker of neuronal injury ATF3 (Fig. 1J). Moreover, TLR8 was co-localized with the non-peptidergic marker IB4, and with the peptidergic marker CGRP, but rarely with the large-diameter neuronal marker NF200 (Fig. 1K–M). These data suggest that TLR8 is mainly expressed in small-diameter neurons in the TG and increases after pIONL.

pIONL causes mechanical allodynia of the ipsilateral facial skin, and increases TLR8 expression in the TG. A Time-course of facial nocifensive behavior score stimulated by a 0.02 g von Frey filament in sham and pIONL mice (**P < 0.01, ***P < 0.001 vs corresponding sham group, two-way RM ANOVA followed by post-hoc Bonferroni test; n = 6 mice/group). B Real-time PCR showing the expression of Tlr8 mRNA in the TG of naïve, sham, and pIONL mice (*P < 0.05, **P < 0.01, ***P < 0.001 vs sham, Student’s t-test; n = 6 mice/group). C–E Representative TLR8 immunofluorescent images showing the distribution of TLR8+ neurons in the TG of naïve (C), sham (D), and pIONL 10 days (E) mice. F Percentages of TLR8+ cells in the TG of naïve, sham, and pIONL mice (***P < 0.001 vs sham, one-way ANOVA followed by the Bonferroni test; n = 3 mice/group). G, H Double immunostaining of TLR8 with Tuj1 (G) and CD68 (H). I Cell-size distribution of TLR8+ neurons and total neurons in the TG 10 days after pIONL. J–M Double-stained immunofluorescent images showing the co-localization of TLR8 with ATF3 (J), IB4 (K), CGRP (L), and NF200 (M) in the TG.
Deletion or Knockdown of Tlr8 in the TG Alleviates pIONL-Induced Mechanical Allodynia
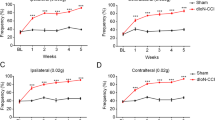
To determine whether TLR8 is involved in TNP, we compared the mechanical allodynia after pIONL in WT and Tlr8−/− mice. As shown in Fig. 2A, the facial nocifensive behavior score evoked by a 0.02 g von Frey filament was significantly decreased from 7 days after pIONL until the end of the observation period in Tlr8−/− mice, suggesting the alleviation of TNP after Tlr8 deletion. To further check if TLR8 in the TG is involved in TNP, we specifically knocked down TLR8 expression in the TG by intra-infraorbital nerve injection of Tlr8 siRNA mixed with RVG-9R [20]. The injection of Tlr8 siRNA at 4 days after pIONL caused a decrease in the nocifensive score. The effect appeared at 3 days after the injection and was maintained for > 48 h (Fig. 2B). RT-PCR showed that Tlr8 siRNA injection decreased the Tlr8 mRNA level by 46.2 ± 14.7% after 24 h (Fig. 2C). These data suggest that TLR8 in the TG is involved in the maintenance of TNP.

Global deletion of Tlr8 or knockdown of Tlr8 in the TG alleviates the mechanical allodynia induced by pIONL. A Nocifensive behavior scores stimulated by a 0.02 g von Frey filament in WT and Tlr8−/− mice (**P < 0.01, ***P < 0.001 vs WT, two-way RM ANOVA followed by the Bonferroni test). B Nocifensive behavior scores stimulated by a 0.02 g von Frey filament after intra-infraorbital nerve injection of Tlr8 siRNA mixed with RVG 4 days after pIONL (***P < 0.001 vs NC siRNA, two-way RM ANOVA followed by the Bonferroni test). C RT-PCR of TG tissue showing that Tlr8 siRNA down-regulates Tlr8 mRNA expression (*P < 0.05 vs NC siRNA, Student’s t-test; n = 5 mice/group).
Deletion of Tlr8 Reduces the pIONL-Induced Activation of ERK and p38, and the Expression of Pro-inflammatory Cytokines in the TG
MAPKs have been well demonstrated in the pathological processes of neuropathic pain [21, 24, 25]. Our previous data showed that ERK and p38, but not JNK are activated in the TG after pIONL [24, 25]. Here we further confirmed the increased expression of pERK and pp38 in the TG 10 days after pIONL in WT mice (Fig. 3A, B). We then compared pERK and pp38 expression in WT and Tlr8−/− mice and found that both pERK and pp38 level in the TG of Tlr8−/− mice were lower than those in WT mice after pIONL (Fig. 3C, D). This suggests that TLR8 is involved in the pIONL-induced activation of ERK and p38 in the TG.

Expression of pERK, pp38, and pro-inflammatory cytokines induced by pIONL is attenuated in the TG of Tlr8−/− mice. A, B Phosphorylation of ERK and p38 is increased at 10 days after pIONL in the TG of WT mice (*P < 0.05, **P < 0.01 vs sham, Student’s t-test; n = 3 mice/group). C, D The phosphorylation of ERK and p38 induced by pIONL is reduced in the TG of Tlr8−/− mice (*P < 0.05, vs WT, Student’s t-test; n = 3 mice/group). E–G The expression of TNF-α (E), IL-1β (F), and IL-6 (G) is increased by pIONL in the TG of WT mice. The fold-increase of TNF-α (E), IL-1β (F), and IL-6 (G) induced by pIONL is lower in Tlr8−/− mice than in WT mice (*P < 0.05, **P < 0.01, ***P < 0.001, pIONL vs sham; #P < 0.05, Tlr8−/− vs WT-pIONL, Student’s t-test; n = 5–6 mice/group).
The pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 have been demonstrated to play an important role in the development and maintenance of neuropathic pain [3, 20, 31]. Our real-time PCR data showed that pIONL increased the expression of TNF-α, IL-1β, and IL-6 in the injured TG of WT mice (Fig. 3E–G). In Tlr8−/− mice, the average fold-increase of TNF-α, IL-1β, and IL-6 induced by pIONL was significantly lower than that in WT mice (Fig. 3E–G). These data suggest that pIONL-induced pro-inflammatory cytokine expression is partly dependent on TLR8 in the TG.
Intra-TG Injection of TLR8 Agonist VTX-2337 Induces Pain Hypersensitivity
Tlr8 and Tlr7 are highly homologous, and both recognize viral single-stranded RNA. Several agonists including CL075, 3M-003, and R848 activate both TLR8 and TLR7 [32,33,34]. VTX-2337 is a selective and potent agonist of TLR8 [20]. We injected VTX-2337 into the TG to determine its effect on the pain hypersensitivity of the face. The behavioral data showed that intra-TG injection of VTX-2337 dose-dependently (50, 100, and 500 ng) increased the facial nocifensive behavior score (Fig. 4A). The pain hypersensitivity emerged 3 h after the injection and lasted for > 6 h. However, the intra-TG injection of VTX-2337 at 100 ng did not induce pain hypersensitivity in Tlr8−/− mice (Fig. 4B). Despite this, the pain hypersensitivity was still evoked in Tlr7−/− mice (Fig. 4C). These data indicate that TLR8 in the TG is sufficient to induce pain hypersensitivity on the face, and also demonstrate that VTX-2337-induced pain hypersensitivity is dependent on TLR8, not TLR7.

The TLR8 agonist VTX-2337 induces pain hypersensitivity in WT and Tlr7−/− mice, but not in Tlr8−/− mice. A Nocifensive behavior scores stimulated by a 0.02 g von Frey filament after VTX-2337 (10, 100, and 500 ng) or PBS injection into the TG in WT mice (*P < 0.05, **P < 0.01, ***P < 0.001 vs PBS, two-way RM ANOVA followed by Bonferroni test). B Nocifensive behavior scores stimulated by a 0.02 g von Frey filament after VTX-2337 (100 ng) and PBS injection in Tlr8−/− mice. C Nocifensive behavior scores stimulated by a 0.02 g von Frey filament after VTX-2337 (100 ng) and PBS injection in Tlr7−/− mice (**P < 0.01, ***P < 0.001 vs PBS, two-way RM ANOVA followed by Bonferroni test).
TLR8 Agonist VTX-2337 Increases the Ca2+ Concentration in TG Neurons
As an important intracellular second messenger, intracellular Ca2+ ([Ca2+]i) is involved in many Ca2+-dependent intracellular signaling pathways. We checked whether VTX-2337 affects the Ca2+ response in HEK293 cells transfected with control or TLR8 plasmids. The Ca2+ imaging data showed that VTX-2337 had no effect on HEK293 cells transfected with control plasmids (Fig. 5A). However, VTX-2337 dose-dependently increased the F340/380 ratio of HEK293 cells transfected with TLR8 plasmids (Fig. 5B). With an increasing concentration of VTX-2337, the [Ca2+]i also increased (Fig. 5C).

The TLR8 agonist VTX-2337 increases the Ca2+ concentration in HEK293 cells transfected with TLR8 plasmids and in TG neurons. A, B Effect of VTX-2337 on the F340/380 ratio in HEK293 cells transfected with control plasmids (A) or TLR8 plasmids (B). C Statistical data showing that VTX-2337 dose-dependently increases the F340/380 fold ratio in HEK293 cells transfected with TLR8 plasmids. D, E Representative Ca2+ images showing the intracellular Ca2+ activity in TG neurons at baseline (D) and with application of VTX-2337 (E) (arrows, neurons responding to VTX-2337). F Representative curves for the F340/380 ratio in TG neurons treated with VTX-2337 (5 μmol/L) in WT mice. G Representative trace of the F340/380 ratio in a TG neuron treated with VTX-2337 (100 nmol/L, 500 nmol/L, and 5 μmol/L) and KCl (50 mmol/L). H Representative trace of the F340/380 ratio in a TG neuron after three treatments with VTX-2337. KCl (50 mmol/L) is used as a positive control. I Percentages of neurons responsive to different concentrations of VTX-2337. J Fold increase of the F340/380 ratio after treatment with different concentrations of VTX-2337. K Representative curves of the F340/380 ratio in TG neurons in Tlr8−/− mice treated with VTX-2337 (5 μmol/L).
We further examined the effect of VTX-2337 on the [Ca2+]i in primary TG neurons. The data showed that VTX-2337 at 5 μmol/L increased the F340/380 ratio (Fig. 5D–F). VTX-2337 at 500 nmol/L induced a [Ca2+]i increase similar to that at 5 μmol/L, but 100 nmol/L VTX-2337 induced lower response (Fig. 5G). To test whether TG neurons desensitize to the repeated application of VTX-2337, we applied it three times at 500-s intervals, and the F340/380 ratio did not differ among the three applications (Fig. 5H). Statistical data showed that the percentage of responding neurons increased with increasing VTX-2337 concentration (Fig. 5I), and the fold-change of the F340/380 ratio was higher after treatment with either 500 nmol/L or 5 μmol/L than with 100 nmol/L VTX-2337 (Fig. 5J). Furthermore, we checked the Ca2+ response of TG neurons to VTX-2337 in Tlr8−/− mice, and found no fold-increase in the F340/380 ratio after incubation with VTX-2337 (5 μmol/L) in Tlr8−/− mice (Fig. 5K).
TLR8 Agonist VTX-2337 Induces ERK and p38 Activation and Pro-inflammatory Cytokine Expression
To determine whether activating TLR8 can drive the activation of ERK and p38 in the TG, we injected VTX-2337 into the TG and checked the expression of pERK and pp38. Western blot showed that VTX-2337 increased the expression of pERK and pp38 in the TG of WT mice (Fig. 6A, B), but not in Tlr8−/− mice (Fig. 6C, D). These data indicate that the activation of ERK and p38 expression induced by VTX-2337 is dependent on TLR8 in the TG.

The TLR8 agonist VTX-2337-induced upregulation of pERK, pp38, and pro-inflammatory cytokines is reduced in Tlr8−/− mice. A, B After intra-TG injection of VTX-2337, the phosphorylation of ERK and p38 is increased in the TG of WT mice (*P < 0.05, **P < 0.01 vs PBS, Student’s t-test; n = 3 mice/group). C, D The phosphorylation of ERK and p38 in the TG does not increase after intra-TG injection of VTX-2337 in Tlr8−/− mice (P > 0.05 vs PBS, Student’s t-test; n = 3 mice/group). E–G The expression of TNF-α (E), IL-1β (F), and IL-6 (G) is increased by intra-TG injection of VTX-2337. The fold-increase of TNF-α (E), IL-1β (F), and IL-6 (G) induced by VTX-2337 is lower in Tlr8−/− mice than in WT mice (**P < 0.01, ***P < 0.001 vs WT-PBS; #P < 0.05, ##P < 0.01 vs WT-VTX-2337 group, Student’s t-test; n = 5 mice/group).
To check the role of TLR8 in the expression of pro-inflammatory cytokines, we examined TNF-α, IL-1β, and IL-6 expression in the TG after intra-TG injection of VTX-2337 in WT and Tlr8−/− mice. The real-time PCR revealed that VTX-2337 dramatically increased their expression in WT mice (P < 0.01 or 0.001 vs PBS, Student’s t-test), but not in Tlr8−/− mice (P > 0.05 vs PBS, Student’s t-test). The fold-increase of TNF-α, IL-1β, and IL-6 in Tlr8−/− mice induced by VTX-2337 was lower than that in WT mice (Fig. 6E–G), indicating that the VTX-2337-induced upregulation of pro-inflammatory cytokines is dependent on TLR8.
Discussion
In this study, we showed that pIONL induced a persistent increase of TLR8 expression in small TG neurons, and the pIONL-induced pain hypersensitivity was alleviated in Tlr8−/− mice and in WT mice treated with Tlr8 siRNA in the TG. In addition, the pIONL-induced neuroinflammation, displayed as MAPK activation and pro-inflammatory cytokine production in the TG, was reduced in Tlr8−/− mice. Consistently, the intra-TG injection of the TLR8 agonist VTX-2337 led to facial pain hypersensitivity, the activation of ERK and p38, and an increase of pro-inflammatory cytokine expression. These results suggest that TLR8 in TG neurons plays a critical role in the maintenance of TNP.
TLR8 is Upregulated in IB4+ and CGRP+ TG Neurons After pIONL and Contributes to the Maintenance of TNP
TLRs, as one type of innate receptors, can recognize microbial pathogens and play an essential role in the initiation of innate immune responses [16]. In recent years, the role of TLRs in pain and itch has been widely investigated [17, 18, 35]. Several TLRs have been demonstrated to be expressed in glial cells and neurons in the DRG under chronic pain conditions [19, 36]. For example, TLR9 is expressed in DRG macrophages and contributes to chemotherapy-induced neuropathic pain [19]. TLR3 is expressed in small TRPV1-positive DRG neurons and regulates sensory neuronal excitability [37]. TLR7 is distributed in TRPA1-positive DRG neurons and is involved in both pain and itch [18, 38, 39]. Although it had been reported that TLR8 is non-functional in mice [40], increasing evidence has shown that TLR8 in immunocytes and neural cells has effects on inflammation and neuronal apoptosis [40, 41]. Our previous data showed that TLR8 is expressed in small IB4-positive DRG neurons and contributes to spinal nerve injury-induced neuropathic pain [20, 42].
In contrast to well-studied TLRs in the DRG underlying chronic pain, the expression of TLRs in the TG and their role in TNP are less studied. Here, we found that TLR8 had low expression in naïve mice but this dramatically increased in pIONL-operated mice. In addition, TLR8 was expressed in both IB4+ and CGRP+ small TG neurons after pIONL. As TLR8 was mainly expressed in IB4+ neurons in the DRG of naïve mice [20], we speculate that pIONL increases TLR8 in IB4+ neurons and induces TLR8 in CGRP+ neurons. Considering that IB4+ and CGRP+ neurons extend C- and Aβ-afferent fibers in the peripheral nervous system, and are widely recognized in the transmission of noxious signals to the spinal cord [20, 43], TLR8 may be involved in processing pain signals. Our behavioral data further showed that Tlr8−/− mice suffered less mechanical allodynia than WT mice, and knockdown of TLR8 in the TG alleviated the TNP in the late phase (> 7 days), not in the earlier phase (< 3 days), suggesting the involvement of TLR8 in the maintenance of TNP. As TLR8 is expressed in primary afferents in the spinal cord [20], we do not exclude a possible role of TLR8 in the medulla oblongata in mediating TNP.
TLR8 Contributes to the Maintenance of TNP via ERK and p38 Activation in the TG
In immunocytes, the canonical downstream signaling of TLR8 in endosomal compartments is associated with the production of cytokines mediated by the NF-κB pathway [17, 36]. However, the signaling pathway of NF-κB in peripheral sensory neurons cannot be activated by the TLR8 agonist VTX-2337 [20]. Instead, VTX-2337 induces ERK activation in the DRG after intrathecal injection, and spinal nerve ligation-induced ERK activation is also reduced in Tlr8−/− mice, suggesting ERK as a downstream signal of TLR8 in the DRG [20]. It has been shown that ligation of the ION induces the activation of ERK and p38, but not JNK in the TG [24, 25]. ERK is also activated in TG neurons induced by migraine or lingual nerve crush [24, 44, 45]. We showed that the pIONL-induced ERK activation was reduced in Tlr8−/− mice. Consistent with this, the intra-TG injection of VTX-2337 also induced pain hypersensitivity and pERK action in WT mice, but not in Tlr8−/− mice. Similarly, the activation of p38 induced by pIONL was decreased in Tlr8−/− mice, and p38 was also activated by VTX-2337 in a TLR8-dependent manner. Given that the inhibition of ERK activation by PD98059 (an MEK inhibitor) attenuates pIONL-induced mechanical allodynia and lingual nerve crush-induced pain hypersensitivity [24, 45], and the intra-TG administration of the p38 inhibitor SB203580 alleviates tongue pain and pIONL-induced TNP [25, 46], TLR8 may contribute to the maintenance of TNP via activation of ERK and p38 in the TG.
TLR8 is Involved in Neuroinflammation
Neuroinflammation, which is characterized by the production of cytokines and chemokines in the nervous system, is one of the hallmarks in neuropathic pain [2, 3, 6, 7, 23, 47,48,49]. Several studies have shown increased levels of the cytokines TNF-α, IL-1β, and IL-6 in the DRG of animals with chronic pain induced by chemotherapeutic paclitaxel, chronic constriction of the sciatic nerve, experimental autoimmune encephalomyelitis, operation, or inflammation [22, 50,51,52]. The decreasing expression of TNF-α, IL-1β, and IL-6 in the DRG alleviates neuropathic and inflammatory pain [48, 51, 52]. Intra-TG injection of the TNF-α inhibitor etanercept, or the IL-1β inhibitor diacerein attenuate the pIONL-induced mechanical allodynia for up to 6 h [24]. In the present study, pIONL or intra-TG injection of VTX-2337 induced an increase of TNF-α, IL-1β, and IL-6 expression in the TG, and this was impeded in Tlr8−/− mice, indicating that TLR8 is upstream of the pro-inflammatory cytokine production in the TG after pION.
Our previous studies showed that inhibition of ERK with PD98059 decreases the pIONL-induced upregulation of TNF-α and IL-1β in the TG [24]. The p38-MAPK inhibitor SB203580 also has a similar effect [25]. Therefore, activation of ERK and p38 in the TG may trigger the production of pro-inflammatory cytokines to induce TNP. Vice versa, pro-inflammatory cytokines activate the MAPK pathway to induce pain hypersensitivity in peripheral sensory neurons. In the DRG, IL-6 activates ERK signaling to induce pain hypersensitivity [53]. Likely, IL-1β also induces pain hypersensitivity through activating p38 in the DRG [54, 55]. Regional application of TNF-α at a nerve root causes pain hypersensitivity and increases pERK in small and medium DRG neurons [56]. Therefore, the activation of MAPKs induces the production of pro-inflammatory cytokines, which conversely boosts activation of the MAPK pathway in the peripheral sensory ganglia, such as the TG and DRG. This whole process creates a vicious cycle for neuropathic pain.
TLR8 is Involved in the Increase of Intracellular Ca2+ and may Enhance Neuronal Excitability
Intracellular Ca2+ affects a wide variety of neuronal responses, including excitability. The excitability of primary sensory neurons increases after peripheral nerve injury [2, 23, 43]. Using Ca2+ imaging, we found that the TLR8 agonist VTX-2337 rapidly increased the [Ca2+]i in HEK293 cells transfected with TLR8 plasmids and primary cultured TG neurons. The increased [Ca2+]i may come from extracellular Ca2+ influx or intracellular Ca2+ release from organelles, such as the ER and lysosomes [10, 39, 57,58,59,60]. TLR8 is localized in the ER, endosomes, and lysosomes in human monocytes and macrophages [61, 62]. We recently demonstrated that TLR8 is also distributed in the same organelles in DRG neurons. Thus, we speculate that the increased Ca2+ after VTX-2337 treatment is released from intracellular stores. But how TLR8 mediates Ca2+ release needs further investigation.
It has been demonstrated that an increase of [Ca2+]i can trigger a myriad of intracellular signals which include the phosphorylation of MAPKs (such as ERK and p38), inflammatory responses, and the release of neurotransmitters [57, 63, 64]. In addition, the activated ERK increases the expression of the Na+ channel NaV1.8, and regulates the TRPV1 activity in peripheral sensory neurons [65, 66]. Similarly, activated p38 also directly regulates the phosphorylation of NaV1.8 channels and increases the level of TRPV1 in DRG neurons [65, 67]. Furthermore, pro-inflammatory cytokines upregulate the expression of NaV1.7 and NaV1.6, further contributing to peripheral sensitization and neuropathic pain [68, 69]. Therefore, the activation of TLR8 may increase the excitability of TG neurons via regulating the expression and function of Na+ channels.
Conclusion
In the present study, we demonstrated that pIONL increased TLR8 expression in small TG neurons. TLR8 may facilitate peripheral sensitization and trigeminal neuropathic pain through increasing intracellular Ca2+, activating MAPKs, increasing pro-inflammatory cytokine expression, and enhancing neuronal excitability. Thus, targeting TLR8 and its downstream signals in the TG may be a novel strategy for the treatment of TNP.
References
Maarbjerg S, Di Stefano G, Bendtsen L, Cruccu G. Trigeminal neuralgia - diagnosis and treatment. Cephalalgia 2017, 37: 648–657.
Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354: 572–577.
Zhang ZJ, Jiang BC, Gao YJ. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell Mol Life Sci 2017, 74: 3275–3291.
Wu XB, He LN, Jiang BC, Wang X, Lu Y, Gao YJ. Increased CXCL13 and CXCR5 in anterior cingulate cortex contributes to neuropathic pain-related conditioned place aversion. Neurosci Bull 2019, 35: 613–623.
Chen G, Luo X, Qadri MY, Berta T, Ji RR. Sex-dependent glial signaling in pathological pain: Distinct roles of spinal microglia and astrocytes. Neurosci Bull 2018, 34: 98–108.
Matsuda M, Huh Y, Ji RR. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J Anesth 2019, 33: 131–139.
Miller RJ, Jung H, Bhangoo SK, White FA. Cytokine and chemokine regulation of sensory neuron function. Handb Exp Pharmacol 2009: 417–449.
Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 2018, 129: 343–366.
Wang ZC, Li LH, Bian C, Yang L, Lv N, Zhang YQ. Involvement of NF-kappaB and the CX3CR1 signaling network in mechanical allodynia induced by tetanic sciatic stimulation. Neurosci Bull 2018, 34: 64–73.
Liu Q, Chen W, Fan X, Wang J, Fu S, Cui S, et al. Upregulation of interleukin-6 on Cav3.2 T-type calcium channels in dorsal root ganglion neurons contributes to neuropathic pain in rats with spinal nerve ligation. Exp Neurol 2019, 317: 226–243.
Zhu D, Fan T, Huo X, Cui J, Cheung CW, Xia Z. Progressive increase of inflammatory CXCR4 and TNF-alpha in the dorsal root ganglia and spinal cord maintains peripheral and central sensitization to diabetic neuropathic pain in rats. Mediators Inflamm 2019, 2019: 4856156.
Zhang J, Su YM, Li D, Cui Y, Huang ZZ, Wei JY, et al. TNF-alpha-mediated JNK activation in the dorsal root ganglion neurons contributes to Bortezomib-induced peripheral neuropathy. Brain Behav Immun 2014, 38: 185–191.
Abdelsadik A, Trad A. Toll-like receptors on the fork roads between innate and adaptive immunity. Hum Immunol 2011, 72: 1188–1193.
Savva A, Roger T. Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front Immunol 2013, 4: 387.
Chiu IM. Infection, Pain, and Itch. Neurosci Bull 2018, 34: 109–119.
Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol 2006, 7: 131–137.
Liu T, Gao YJ, Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull 2012, 28: 131–144.
He L, Han G, Wu S, Du S, Zhang Y, Liu W, et al. Toll-like receptor 7 contributes to neuropathic pain by activating NF-kappaB in primary sensory neurons. Brain Behav Immun 2020, 87: 840–851.
Luo X, Huh Y, Bang S, He Q, Zhang L, Matsuda M, et al. Macrophage Toll-like receptor 9 contributes to chemotherapy-induced neuropathic pain in male mice. J Neurosci 2019, 39: 6848–6864.
Zhang ZJ, Guo JS, Li SS, Wu XB, Cao DL, Jiang BC, et al. TLR8 and its endogenous ligand miR-21 contribute to neuropathic pain in murine DRG. J Exp Med 2018, 215: 3019–3037.
Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev 2009, 60: 135–148.
Gu HW, Xing F, Jiang MJ, Wang Y, Bai L, Zhang J, et al. Upregulation of matrix metalloproteinase-9/2 in the wounded tissue, dorsal root ganglia, and spinal cord is involved in the development of postoperative pain. Brain Res 2019, 1718: 64–74.
Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov 2014, 13: 533–548.
Zhang Q, Cao DL, Zhang ZJ, Jiang BC, Gao YJ. Chemokine CXCL13 mediates orofacial neuropathic pain via CXCR5/ERK pathway in the trigeminal ganglion of mice. J Neuroinflammation 2016, 13: 183.
Zhang Q, Zhu MD, Cao DL, Bai XQ, Gao YJ, Wu XB. Chemokine CXCL13 activates p38 MAPK in the trigeminal ganglion after infraorbital nerve injury. Inflammation 2017, 40: 762–769.
Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lu N, et al. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J Clin Invest 2014, 124: 1173–1186.
Neubert JK, Mannes AJ, Keller J, Wexel M, Iadarola MJ, Caudle RM. Peripheral targeting of the trigeminal ganglion via the infraorbital foramen as a therapeutic strategy. Brain Res Brain Res Protoc 2005, 15: 119–126.
Zhang Y, Chen Y, Liedtke W, Wang F. Lack of evidence for ectopic sprouting of genetically labeled Abeta touch afferents in inflammatory and neuropathic trigeminal pain. Mol Pain 2015, 11: 18.
Kernisant M, Gear RW, Jasmin L, Vit JP, Ohara PT. Chronic constriction injury of the infraorbital nerve in the rat using modified syringe needle. J Neurosci Methods 2008, 172: 43–47.
Lee JH, Park CK, Chen G, Han Q, Xie RG, Liu T, et al. A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell 2014, 157: 1393–1404.
Xu Y, Jiang Y, Wang L, Huang J, Wen J, Lv H, et al. Thymosin alpha-1 inhibits complete freund’s adjuvant-induced pain and production of microglia-mediated pro-inflammatory cytokines in spinal cord. Neurosci Bull 2019, 35: 637–648.
Spranger S, Javorovic M, Burdek M, Wilde S, Mosetter B, Tippmer S, et al. Generation of Th1-polarizing dendritic cells using the TLR7/8 agonist CL075. J Immunol 2010, 185: 738–747.
Gorden KK, Qiu X, Battiste JJ, Wightman PP, Vasilakos JP, Alkan SS. Oligodeoxynucleotides differentially modulate activation of TLR7 and TLR8 by imidazoquinolines. J Immunol 2006, 177: 8164–8170.
Talreja J, Samavati L. K63-linked polyubiquitination on TRAF6 regulates LPS-mediated MAPK activation, cytokine production, and bacterial clearance in Toll-like receptor 7/8 primed murine macrophages. Front Immunol 2018, 9: 279.
Ji RR. Neuroimmune interactions in itch: Do chronic itch, chronic pain, and chronic cough share similar mechanisms? Pulm Pharmacol Ther 2015, 35: 81–86.
Liu XJ, Liu T, Chen G, Wang B, Yu XL, Yin C, et al. TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Sci Rep 2016, 6: 28188.
Liu T, Berta T, Xu ZZ, Park CK, Zhang L, Lu N, et al. TLR3 deficiency impairs spinal cord synaptic transmission, central sensitization, and pruritus in mice. J Clin Invest 2012, 122: 2195–2207.
Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Toll-like receptor 7 mediates pruritus. Nat Neurosci 2010, 13: 1460–1462.
Park CK, Xu ZZ, Berta T, Han Q, Chen G, Liu XJ, et al. Extracellular microRNAs activate nociceptor neurons to elicit pain via TLR7 and TRPA1. Neuron 2014, 82: 47–54.
Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol 2002, 3: 499.
Tang SC, Yeh SJ, Li YI, Wang YC, Baik SH, Santro T, et al. Evidence for a detrimental role of TLR8 in ischemic stroke. Exp Neurol 2013, 250: 341–347.
Wang ZH, Liu T. MicroRNA21 meets neuronal TLR8: Non-canonical functions of MicroRNA in neuropathic pain. Neurosci Bull 2019, 35: 949–952.
Berta T, Qadri Y, Tan PH, Ji RR. Targeting dorsal root ganglia and primary sensory neurons for the treatment of chronic pain. Expert Opin Ther Targets 2017, 21: 695–703.
Hawkins JL, Moore NJ, Miley D, Durham PL. Secondary traumatic stress increases expression of proteins implicated in peripheral and central sensitization of trigeminal neurons. Brain Res 2018, 1687: 162–172.
Mikuzuki L, Saito H, Katagiri A, Okada S, Sugawara S, Kubo A, et al. Phenotypic change in trigeminal ganglion neurons associated with satellite cell activation via extracellular signal-regulated kinase phosphorylation is involved in lingual neuropathic pain. Eur J Neurosci 2017, 46: 2190–2202.
Maruno M, Shinoda M, Honda K, Ito R, Urata K, Watanabe M, et al. Phosphorylation of p38 in trigeminal ganglion neurons contributes to tongue heat hypersensitivity in mice. J Oral Facial Pain Headache 2017, 31: 372–380.
Huh Y, Ji RR, Chen G. Neuroinflammation, bone marrow stem cells, and chronic pain. Front Immunol 2017, 8: 1014.
Zhu MD, Zhao LX, Wang XT, Gao YJ, Zhang ZJ. Ligustilide inhibits microglia-mediated proinflammatory cytokines production and inflammatory pain. Brain Res Bull 2014, 109: 54–60.
Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Armijo LM, et al. Intrathecal cannabilactone CB(2)R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain 2012, 153: 1091–1106.
Matsuoka Y, Yamashita A, Matsuda M, Kawai K, Sawa T, Amaya F. NLRP2 inflammasome in dorsal root ganglion as a novel molecular platform that produces inflammatory pain hypersensitivity. Pain 2019, 160: 2149–2160.
Zhang X, Jiang N, Li J, Zhang D, Lv X. Rapamycin alleviates proinflammatory cytokines and nociceptive behavior induced by chemotherapeutic paclitaxel. Neurol Res 2019, 41: 52–59.
Zychowska M, Rojewska E, Makuch W, Luvisetto S, Pavone F, Marinelli S, et al. Participation of pro- and anti-nociceptive interleukins in botulinum toxin A-induced analgesia in a rat model of neuropathic pain. Eur J Pharmacol 2016, 791: 377–388.
Tillu DV, Melemedjian OK, Asiedu MN, Qu N, De Felice M, Dussor G, et al. Resveratrol engages AMPK to attenuate ERK and mTOR signaling in sensory neurons and inhibits incision-induced acute and chronic pain. Mol Pain 2012, 8: 5.
Eijkelkamp N, Heijnen CJ, Carbajal AG, Willemen HL, Wang H, Minett MS, et al. G protein-coupled receptor kinase 6 acts as a critical regulator of cytokine-induced hyperalgesia by promoting phosphatidylinositol 3-kinase and inhibiting p38 signaling. Mol Med 2012, 18: 556–564.
Zelenka M, Schafers M, Sommer C. Intraneural injection of interleukin-1beta and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain 2005, 116: 257–263.
Takahashi N, Kikuchi S, Shubayev VI, Campana WM, Myers RR. TNF-alpha and phosphorylation of ERK in DRG and spinal cord: insights into mechanisms of sciatica. Spine (Phila Pa 1976) 2006, 31: 523–529.
Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, Zamponi GW. Calcium-permeable ion channels in pain signaling. Physiol Rev 2014, 94: 81–140.
Rozas P, Lazcano P, Pina R, Cho A, Terse A, Pertusa M, et al. Targeted overexpression of tumor necrosis factor-alpha increases cyclin-dependent kinase 5 activity and TRPV1-dependent Ca2+ influx in trigeminal neurons. Pain 2016, 157: 1346–1362.
Haustrate A, Prevarskaya N, Lehen’kyi V. Role of the TRPV channels in the endoplasmic reticulum calcium homeostasis. Cells 2020, 9: 317. https://doi.org/10.3390/cells9020317.
Terashima R, Kimura M, Higashikawa A, Kojima Y, Ichinohe T, Tazaki M, et al. Intracellular Ca(2+) mobilization pathway via bradykinin B1 receptor activation in rat trigeminal ganglion neurons. J Physiol Sci 2019, 69: 199–209.
Itoh H, Tatematsu M, Watanabe A, Iwano K, Funami K, Seya T, et al. UNC93B1 physically associates with human TLR8 and regulates TLR8-mediated signaling. PLoS One 2011, 6: e28500.
Ishii N, Funami K, Tatematsu M, Seya T, Matsumoto M. Endosomal localization of TLR8 confers distinctive proteolytic processing on human myeloid cells. J Immunol 2014, 193: 5118–5128.
Canzobre MC, Paganelli AR, Rios H. Effect of periapical inflammation on calcium binding proteins and ERK in the trigeminal nucleus. Acta Odontol Latinoam 2019, 32: 103–110.
Wang H, Wei Y, Pu Y, Jiang D, Jiang X, Zhang Y, et al. Brain-derived neurotrophic factor stimulation of T-type Ca(2+) channels in sensory neurons contributes to increased peripheral pain sensitivity. Sci Signal 2019, 12(600): eaaw2300. https://doi.org/10.1126/scisignal.aaw2300.
Hudmon A, Choi JS, Tyrrell L, Black JA, Rush AM, Waxman SG, et al. Phosphorylation of sodium channel Na(v)1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci 2008, 28: 3190–3201.
Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci 2004, 24: 8300–8309.
Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 2002, 36: 57–68.
Ding HH, Zhang SB, Lv YY, Ma C, Liu M, Zhang KB, et al. TNF-alpha/STAT3 pathway epigenetically upregulates NaV1.6 expression in DRG and contributes to neuropathic pain induced by L5-VRT. J Neuroinflammation 2019, 16: 29.
Tamura R, Nemoto T, Maruta T, Onizuka S, Yanagita T, Wada A, et al. Up-regulation of NaV1.7 sodium channels expression by tumor necrosis factor-alpha in cultured bovine adrenal chromaffin cells and rat dorsal root ganglion neurons. Anesth Analg 2014, 118: 318–324.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31970938, 31671091, 31871064, 81571070, and 31700899), and the Natural Science Research Program of Jiangsu Province, China (BK20171255 and BK20191448), the Qing Lan Project, and the Innovation and Entrepreneurship Training Program for College Students in Jiangsu Province, China (201810304029Z).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, LX., Jiang, M., Bai, XQ. et al. TLR8 in the Trigeminal Ganglion Contributes to the Maintenance of Trigeminal Neuropathic Pain in Mice. Neurosci. Bull. 37, 550–562 (2021). https://doi.org/10.1007/s12264-020-00621-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-020-00621-4