
Abstract
The requirement of systems biology for connecting different levels of biological research leads directly to a need for integrating vast amounts of diverse information in general and of omics data in particular. The nutritional phenotype database addresses this challenge for nutrigenomics. A particularly urgent objective in coping with the data avalanche is making biologically meaningful information accessible to the researcher. This contribution describes how we intend to meet this objective with the nutritional phenotype database. We outline relevant parts of the system architecture, describe the kinds of data managed by it, and show how the system can support retrieval of biologically meaningful information by means of ontologies, full-text queries, and structured queries. Our contribution points out critical points, describes several technical hurdles. It demonstrates how pathway analysis can improve queries and comparisons for nutrition studies. Finally, three directions for future research are given.
Similar content being viewed by others


Introduction
Biology is rapidly developing into a data-driven science and faces not only the challenge of coping with an ever growing amount of data but also that of interpreting its complex diversity. The challenge is particularly urgent in domains that need to integrate multi-omics data [4, 5]. This contribution investigates how user interaction within the nutritional phenotype database (dbNP) [18] can be implemented to meet the demands of modern nutrition research for retrieving biologically meaningful data from a multi-omics database.
The remainder of this article is organized as follows. “The nutritional phenotype database” section describes the dbNP. The application of controlled vocabulary and ontologies is presented in “Controlled vocabularies and ontologies” section. “Organization of data in the dbNP” section introduces the different kinds of data distinguished in the dbNP. “Queries and comparisons” section explains querying in the dbNP. In “Pathway analysis for omics profiles” section, a use case on pathway analysis demonstrates how additional functionality can extend the scope of the dbNP. “Conclusion” section concludes this article.
The nutritional phenotype database
When fully developed, the nutritional phenotype database, (dbNP, van Ommen et al. [18]) will be a research and collaboration tool and a publicly available data and knowledge repository for nutrition-related research. The dbNP aims at being an extensive systems biology framework for nutrition research that in particular facilitates storage and retrieval of a range of relevant kinds of data such as preprocessed omics data, phenotype data, and study metadata. The biological information managed by the dbNP includes data from genetics, transcriptomics, proteomics, simple assay data, metabolomics, functional assays, food intake, and food composition and is tailored to the needs of nutrition research. The raw information and preprocessed data are embedded in an environment of standardized procedures and protocols, annotations, networking, and integrated bioinformatics. The dbNP is an evolving enterprise aiming at acceptance by a wide nutrition and health research community as an open source, precompetitive, and publicly available resource (cf. http://www.dbnp.org). It involves several consortia and organizations; currently the most active ones are Nutrigenomics Organization (NuGO), the Netherlands Metabolomics Centre (NMC), The Netherlands Bioinformatics Centre (NBIC), Eurreca, and The Nutrigenomics Consortium (NGC).
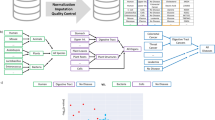
To facilitate the complex functionality, the dbNP is realized in a modular architecture. Each module contains specific data or functionality to operate on special data (cf. Fig. 1). For instance, the transcriptomics module stores raw data from assays used in nutrition studies and also preprocesses and organizes the data for retrieval in combination with other modules. The Simple Assay Module contains all clinical measurement data and units, reference values, and uniform terminology, for referring to measurements such as cholesterol levels in blood samples, or weights of liver samples. Similarly, the metabolomics data stores its raw data as peak tables, which in processed form lead to metabolite concentrations.

Schematic overview of the dbNP with generic study capturing framework (GSF), simple assay, transcriptomics, and metabolomics module
The Generic Study Capturing Framework (GSCF) is a module that provides the user with the means of directly accessing the dbNP by allowing to input and retrieve nutrition studies. In particular, the GSCF aims at supporting the MAGE-TAB [11] and ISA-TAB [12] formats for annotating and communicating microarray data. The study metadata are either imported from existing formats or are input by the user directly. A flexible templating system makes input re-usable and helps reducing the tediousness of the input task. The actual assays containing the data are imported.
Controlled vocabularies and ontologies
Ontologies are formal representations of the knowledge within a domain as a set of concepts and relationships between those concepts [6]. Ontologies define a shared vocabulary, i.e., a standardized set of terminology that eliminates redundancies for common usage within a community. In biology, ontologies have long been employed in the form of taxonomies. Bioinformatics relies on ontologies as standard tools for controlling vocabulary [9, 13] which offer the advantage of centrally managed terminologies. A common purpose of ontologies is to guarantee the long-term uniformity of data entering a system from a variety of sources. Two approaches exist serving this goal. In the first approach, controlled vocabularies enforce standardized input prohibiting any entries not matching known classes in the ontology. However, this may be seen as restrictive and overtly limiting from the user’s perspective. Moreover, the ontology must be left open for adjustment in order to cope with possible gaps in the vocabulary. In the alternative approach, ontologies map ambiguous input automatically onto a unique identifier to indicate that synonyms refer to the same entity. In practice, the latter automated approach suffers from the word sense disambiguation problem which remains an ongoing challenge [1].
An example of employing controlled vocabulary in the GSCF is the species selection in the metadata which describes a study. The user selects a species in a text box that supports autocompletion for the species known to the ontology. After entering for instance mouse as the species, a mouse strain ontology is used next. The current implementation of the dbNP uses ontologies to control vocabulary. Beyond that, ontologies may in the future be linked to automatically supply additionally relevant information. For instance, a chemistry ontology could supply information on chemical compounds, e.g., a drug. The ontology could access information on the composition of the drug and present it to the user or allow automated inferences on its biochemical properties for information retrieval.
The GSCF employs the ontology web services curated by the BioPortal of the National Center for Biomedical Ontology (NCBO) [9] which grants access to hundreds of specialized ontologies on a wide range of domains such as anatomy, phenotype, experimental conditions, imaging, chemistry, and health to provide data integration, information retrieval, and decision support. The control of vocabulary in the dbNP is provided by OBO ontologies [14]. BioPortal already supports the Resource Description Format (RDF). This standard promoted by the Semantic Web Interest Group aims at allowing semantic annotations for web services which may enable the dbNP to execute automatic inferences using the relationships of complex ontologies represented by the semantic web (Table 1).
Organization of data in the dbNP
The complexity of nutrition research captured by the dbNP in general and the diversity of omics data in general require us to distinguish between different kinds of data encountered in nutrition studies. On the highest level of abstraction, study metadata are separated from study data. Study metadata consists of all information that is available before a study is conducted. It may contain the study’s principal investigator, the date the study commenced, the species on which an experiment was conducted, the number of subjects and their organization in groups, the protocols executed for treatment and sampling, ethical statements, a summary description of the study, etc.
As opposed to study metadata, study data are the actual data produced in a study. We note that the distinction between study metadata and study data often leaves room for a gray zone, for instance when the weight of subjects is registered before the study commences. In this case, the initial weight of each subject may be part of the study metadata or of the study data.
Study data can be further partitioned into four subkinds of data as follows. (1) Raw data are the data measured in the respective omics field as produced by the measuring equipment; (2) clean data are the data that results from preprocessing raw data and after quality control and removal of technical variation as far as possible; (3) result data are generated by statistical analysis of the clean data; (4) profiles are data obtained from further processing result data for the purpose of comparing different studies or features with respect to particular kinds of analysis that are not necessarily part of the original study. The four kinds of study data are produced sequentially within an omics module by consecutive processing.
We illustrate the fourfold distinction by considering the transcriptomics module. In a transcriptomics study, the data collected is microarray data. The raw data of the transcriptomics module may consist for instance of Affymetrix CEL files that are stored or linked into the dbNP from an external repository. The clean data consists of gene expression values which result from the raw data after quality control and several steps of normalization and filtering. The result data of a transcriptomics study are identified regulations of genes based on statistical evaluation of clean data in groups of subjects. The result data may be represented by fold changes or p-values of an ANOVA. The profile of a transcriptomics study may include the results of pathway or Gene Ontology [2, 15] analysis performed on the clean data. We remark that the four kinds of data need not all be present in one study. For instance, the weight of a subject is simple assay data (and thus a “non-omics” measurement) that does not require preprocessing, and there may be categorized as clean data.
Queries and comparisons
Querying the dbNP is the interactive process of accessing biologically relevant data across the categories of data introduced in “Organization of data in the dbNP” section. In this section, we briefly describe the relation between the four kinds of study data and three types of user interaction for retrieving study data: (1) full-text queries, (2) structured queries, and (3) study comparisons.
A full-text query on the study-meta data of all studies in the dbNP is the most basic type of query that searches study metadata for a specific string of text. We note that full-text search can also be applied to other Modules. For instance, a keyword like “cholesterol” can return a hit if found in the description of assay data in the Simple Assay Module. The result of a full-text search is a list of short descriptions of all studies that contain the searched text. A full-text query may be convenient for retrieving all studies conducted by the same principal investigator or all studies produced by an institution in a certain year. There are three obvious drawbacks concerning the full-text query. First, it is easy to miss data by not knowing the exact text to search for. Second, study result data are not taken into account. Third, the query may often be too general and produce too many matching studies. The latter problem can be slightly amended by ranking the search results. Accuracy of the text match and other criteria such as time constraints on the data specified by the user can make the problem easier. The first problem can be tackled to a large extend by also using ontologies during the query stage. This approach is used in dbNP. For instance, a drop-down list restricts querying to studies conducted on a specific species. The list is generated by an ontology and excludes improper user input.
dbNP also addresses these drawbacks by providing structured queries on study data. In a structured query, the user specifies feature value pairs for a specific kind of clean data depending on the omics- module the data are stored in. A crucial reason for allowing structured querying is that the data are already organized in modules and partially structured by the database schema of dbNP. For instance, a researcher interested in finding results related to a specific fat treatment can submit a query directly to the Simple Assay Module. Similarly, a query covers a list of compounds with certain values and a treatment with a specific dosage, e.g., 1 microgram of Vitamin B12. Structured queries may combine study metadata and clean data, e.g., to retrieve all studies in which rats of a certain strain were treated with a glucose challenge and produced samples in which a clinical marker was present above a certain level. Queries may access result data. For instance, identifying studies that contain transcriptomics data records with a change in the expression in a specific gene is possible.
Study comparisons not only allow retrieval of studies with similar results but also allow interactive and more in-depth investigation of the studies that result from other queries. This is achieved by a synoptic view of the studies in which similar data fields are juxtaposed. The comparison may enforce recalculation of some of the clean data, for instance, to re-calculate gene regulations for only a particular subgroup of test subjects.
Pathway analysis for omics profiles
Identifying biologically meaningful information in the vastness of the collective noisy data provided by all microarray experiments is an ongoing challenge for database systems in the field of transcriptomics. Pathway analysis is one way of addressing the challenge of improving the interpretation of microarray data. Instead of considering single genes, pathway analysis aims at identifying networks of genes and regulation of genes within such networks [3]. This section illustrates how pathway analysis improves query-related functionality in the transcriptomics module with respect to three items: (1) structured querying, (2) study comparisons, and (3) module-wide analysis of transcriptomics data. The results of this demonstration can also be extrapolated to the other omics modules like those for metabolomics, proteomics, epigenomics, and ChIP studies. The demonstration also shows the feasibility of this approach which will be integrated into the dbNP.
In structured querying, a single pathway or a list of pathways can be entered in the transcriptomics section in order to search for studies in which a pathway is regulated instead of a gene. The corresponding gene identifiers are entered together with the kind of regulation (up, down, arbitrary) and the strength of regulation (e.g., the fold change level) of interest. In order to supply an answer to such a query, the query engine needs to rely on profile data for the transcriptomics module which is either computed on-line, precomputed, or cached. The query results in a list of studies in which the sought after regulation occurs.
Pathway analysis can be performed in relation to dbNP by connecting to PathVisio [16] through web services. This allows taking into account the latest pathway data available from WikiPathways. PathVisio is an open source software tool for displaying and editing pathways. It gives biologically meaningful interpretations of pathways by supplying links from pathway elements to biological entities such as genes or proteins. Moreover, PathVisio supplies a set of analysis tools as plugins. In particular, it can analyze microarray data or other gene-related omics data by mapping genes identified in a microarray to pathways stored in pathway repositories. A variety of sources of identifiers for multi-omics can be connected by relying on the BridgeDB web service [17]. Currently, this type of webservice-based pathway analysis can most be easily performed using the WikiPathways webservices [8], which for instance also allows reanalysis of data from ArrayExpress Gene Expression Atlas [7] and will be used in the same way to evaluate data from dbNP.
Pathway analysis can extend the study comparison by including the effects the studies have shown on a particular pathway or list of pathways. Using PathVisio, the effects on the pathway can also be visualized showing differences in regulation by means of color coding and giving additional information such as links to the literature.
Figure 2 shows how two sets of array data are compared. In this case, the data stems from different tissues within the same study. The two subtables represent the pathways in both kinds of tissue (liver on the left-hand, muscle on the right-hand side). The pathways in both tables are sorted by the strength of the regulation. Thus, the Z-scores are decreasing from top to bottom. In Fig. 3, additional information on the pathway for the linoleic acid metabolism is given. This pathway appeared in the liver tissue sample of Fig. 2 in which it ranked fifth in regulation based on its Z score. Figure 3 indicates a list of studies in which the same pathway is regulated. Other liver samples appear among the listed studies indicating potentially related outcomes.

Comparison of pathway regulation on two kinds of tissue (screenshot of prototype)

Overview: studies with regulation on selected pathway (screenshot of prototype)
Beltrame et al. [3] applied pathway signatures similar to the pathway-related profile data of the dbNP to find similarities between microarray experiments and showed that this approach may produce more reliable results than gene-based approaches. Analogously, dbNP can profit from comparing nutrition studies by a similarity metric based on the pathway profile data. Such a metric could then be applied in clustering or other statistical analysis.
A simple metric could be introduced by comparing lists of the most regulated pathways of two studies. For instance, two studies with heavily up-regulated apoptosis pathway could be considered similar. This idea could be extended further to define a measurement of similarity between more than two pathways. For instance, the ten most regulated pathways in two compared studies could be used to calculate the similarity. An advantage of this metric is that it is computationally cheap if the pathway profile data are stored. The same approach could also be used for GO analysis if results for GO categories are also stored. Essentially these approaches would allow the user to find studies with similar results on a meaningful biological level, and they could even be applied on cross omics comparisons.
Automated analysis of the total profile data, however, also requires careful technical consideration. First, different thresholds for fold changes or p-values for regulation may be of interest depending on the specific research question. Therefore, recalculating pathway profile data may be necessary. Second, in order to maintain comparativeness between all setups the profile data must be calculated according to a uniform procedure. Third, differences in the experimental setup of a study may render a comparison invalid. A nutrition study, for instance, could measure an effect based on the time resulting in microarray data from different time points or using multiple groups of subjects. The data produced by such a setup may be difficult to store in a comparable format that also applies to setups in which the effect is only measured for a specific treatment only once and only to one control group. Fourth, while the same regulation may be found in two pathways, the pattern of regulation within the pathways may still be different. In spite of such difficulties, the similarity metric may still be applied to a large part of the data without much practical constraints resulting in biologically meaningful answers to the biologists questions.
Conclusion
In this contribution, we gave an overview of how biologically relevant information in the nutritional multi-omics framework dbNP can be retrieved interactively. We outlined the dbNP, described in what manner ontologies support user interaction, discussed different kinds of data stored in the system, and showed how interactive retrieval of relevant information is achieved by queries. Furthermore, we discussed an exemplary use case demonstrating the added value and querying of pathway analysis in the transcriptomics module of the dbNP. An automated pathway analysis can be used to show which networks of genes are regulated. With this extra functionality, the user can discover studies in which a certain pathway is regulated. Moreover, the biologist can discover studies in which similar regulations of pathways occur.
Future research will investigate how automated analysis of profile data may support nutrition research. A ranking system for free-text querying will further improve user interaction in the dbNP. Finally, deeper integration of the transcriptomics module with other existing on-line resources will be pursued.
References
Andreopoulos B, Alexopoulou D, Schroeder M (2008) Word sense disambiguation in biomedical ontologies with term co-occurrence analysis and document clustering. Int J Data Min Bioinform 2(3):193–215. doi:10.1504/IJDMB.2008.020522
Baehrecke EH, Dang N, Babaria N, Shneiderman B (2004) Visualization and analysis of microarray and gene ontology data with treemaps. BMC Bioinformatics 5:84. doi:10.1186/1471-2105-5-84
Beltrame L, Rizzetto L, Paola R, Rocca-Serra P, Gambineri L, Battaglia C, Cavalieri D (2009) Using pathway signatures as means of identifying similarities among microarray experiments. PLoS ONE 4(1):4128. doi:10.1371/journal.pone.0004128
Field D, Sansone SA, Collis A, Booth T, Dukes P, Gregurick SK, Kennedy K, Kolar P, Kolker E, Maxon M, Millard S, Mugabushaka AM, Perrin N, Remacle JE, Remington K, Rocca-Serra P, Taylor CF, Thorley M, Tiwari B, Wilbanks J (2009) Megascience ‘Omics data sharing. Science 326(5950):2343–2346
Garrity GM, Field D, Kyrpides N, Hirschman L, Sanson S, Angiuoli S, Cole JR, Glöckner FO, Kolker E, Kowalchuk G, Moran MA, Ussery D, White O (2008) Toward a standards-compliant genomic and metagenomic publication record. OMICS 12(2), Mary Ann Liebert Inc. doi:10.1089
Gruber TR (1998) Toward principles for the design of ontologies used for knowledge sharing. Int J Hum Comput Stud 43(5–6):907–928
Kapushesky M, Emam I, Holloway E, Kurnosov P, Zorin A, Malone J, Rustici G, Williams E, Parkinson H and Brazma A (2010) Gene expression atlas at the european bioinformatics institute. Nucleic Acids Res 38, Database issue D690-D698
Kelder T, Pico AR, Hanspers K, van Iersel MP, Evelo C, Conklin BR (2009) Mining biological pathways using WikiPathways web services. PLoS One 4(7):07. doi:10.1371/journal.pone.0006447
Noy NF, Shah NH, Whetzel PL, Dai B, Dorf M, Griffith N, Jonquet C, Rubin DL, Storey MA, Chute CG, Musen MA (2009) BioPortal: ontologies and integrated data resources at the click of a mouse. Nucleic Acids Res 37:170–173
Pico AR, Kelder T, van Iersel MP, Hanspers K, Conklin BR, Evelo C (2008) WikiPathways: pathway editing for the people. PLoS Biol 6(7). doi:10.1371/journal.pbio.0060184
Rayner TF, Ibne Rezwan F, Lukk M, Zheng Bradley X, Farne A, Holloway E, Malone J, Williams E, Parkinson H (2009) MAGETabulator, a suite of tools to support the microarray data format MAGE-TAB. Bioinformatics 25(2):279–280. doi:10.1093/bioinformatics/btn617
Rocca-Serra P, Brandizi M, Maguire E, Sklyar N, Taylor C, Begley K, Field F, Harris S, Hide W, Hofmann O, Neumann S, Sterk P, Tong W, Sansone SA (2010) ISA software suite: supporting standards-compliant experimental annotation and enabling curation at the community level. Bioinformatics 26(18):2354–2356. doi:10.1093/bioinformatics/btq415
Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Mizrachi I, Ostell J, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Yaschenko E, Ye J (2009) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 37(Database issue):D5–15
Smith B, Ashburner M, Rosse C, Bard J, Bug W, Ceusters W, Goldberg LJ, Eilbeck K, Ireland A, Mungall CJ (2007) The OBO foundry: coordinated evolution of ontologies to support biomedical data integration. In: Nature Biotechnology, vol 25(11). Nature Publishing Group, USA, pp 1251–1255
The Gene Ontology Consortium (2000) Gene ontology: tool for the unification of biology. Nat Genet 25(1):25–29
van Iersel MP, Kelder T, Pico AR, Hanspers K, Coort S, Conklin BR, Evelo C (2008) Presenting and exploring biological pathways with PathVisio. BMC Bioinformatics 9:399
van Iersel MP, Pico AR, Kelder T, Gao J, Ho I, Hanspers K, Conklin BR, Evelo CT (2010) The BridgeDb framework: standardized access to gene, protein and metabolite identifier mapping services. BMC Bioinformatics 11(1):5
van Ommen B, Bouwman J, Dragsted L, Drevon CA, Elliott R, de Groot P, Kaput J, Mathers JC, Muller M, Pepping F, Saito J, Scalbert A, Radonjic M, Rocca-Serra P, Travis T, Wopereis S, Evelo CT (2010) Challenges of molecular nutrition research 6: the nutritional phenotype database to store, share and evaluate nutritional systems biology studies. The nutritional phenotype database to store, share and evaluate nutritional systems biology studies. Genes and Nutrition 5:189–203. doi:10.1007/s12263-010-0167-9
Acknowledgments
We thank Martijn van Iersel and Thomas Kelder for their support with PathVisio and their valuable comments on the text. Financial support for the first development stages of dbNP was given by the European Network of Excellence for Nutrigenomics (NuGO), the Netherlands Metabolomics Centre, and the Netherlands Bioinformatics Centre (NBIC).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Evelo, C.T., van Bochove, K. & Saito, JT. Answering biological questions: querying a systems biology database for nutrigenomics. Genes Nutr 6, 81–87 (2011). https://doi.org/10.1007/s12263-010-0190-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12263-010-0190-x