
Summary
Soft tissue sarcomas are exceedingly rare cancers with more than 100 different histiologic subtypes and tremendous morphologic heterogeneity. Next generation sequencing (NGS) techniques have drastically improved our knowledge about these tumours. The possibility to use these techniques routinely changed the diagnostic standards in soft tissue pathology. Within the last few years pathologists were confronted with a growing number of exceedingly rare, partly molecularly defined, tumour entities. The proposed pathologic diagnosis dictates prognosis and treatment. Therefore, specialized soft tissue pathology units with the possibility to include molecular diagnostics into a histopathological report for diagnostic, predictive and prognostic aspects are needed. A second opinion by a soft tissue pathologist, before final treatment decision, should be mandatory to ensure optimal treatment for all patients by a multidisciplinary team in a specialized centre.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soft tissue sarcomas (STS) are rare malignant tumours arising from connective tissues. The estimated incidence for adult and visceral sarcomas (excluding Gastrointestinal Stromal Tumour, GIST) is 4–5/100,000/year in Europa [1]. STS are a diverse family of malignancies with over 100 histological subtypes (number continuously growing). Most frequent sarcoma subtypes like liposarcoma and leiomyosarcoma have an incidence of <1/100,000/year, whereas the majority of sarcoma histiotypes have an incidence of <1/1,000,000/year [2].
Developments in sarcoma research, mainly driven by new technologies, especially next generation sequencing (NGS) techniques, have drastically improved our knowledge about these rare tumours. The possibility of molecular classification of these tumours has changed the diagnostic standards in pathology. Although these new techniques have facilitated a more reproducible sarcoma classification, correctly diagnosing sarcomas still remains challenging.
Within the last few years pathologists were and are confronted with a growing number of exceedingly rare tumour entities. In part these tumours demonstrate characteristic genetic changes and the use of new molecular techniques, especially NGS, are becoming part of the diagnostic workup of soft tissue tumours. Specialized pathology centres are starting to include molecular diagnostics into their histopathological report for diagnostic, predictive and prognostic aspects.
Despite the recent developments in soft tissue pathology, diagnostic challenges remain and need to be expressed. Knowledge about diagnostic limitations in soft tissue pathology is essential for all physicians treating sarcoma patients.
Diagnostic challenges in sarcoma pathology:
-
The rareness of STS
-
The significant (continuously growing) number of STS as well as the dynamically changing nomenclature of STS
-
The considerable morphological heterogeneity within a soft tissue sarcoma and between different soft tissue sarcomas
-
The morphologic overlap between benign and malignant soft tissue tumours (STT)
-
The trend of limited biopsy material available for pathologic examination and diagnosis of soft tissue tumours
-
Limitations of diagnosing and grading STS on core needle biopsies
Pathology
The tasks of pathology in sarcoma diagnosis is complex, starting by achieving awareness about the need of rapidly placing tumour samples into 4% buffered formalin and to collect fresh frozen tumour tissue (and blood samples) to allow new molecular pathology assessments even at a later stage of disease when requested. Notation of fixation time, the availability of informed consent for bio-banking of patient’s material according to local and international rules will become increasingly important for innovative diagnostics and especially to offer patients the possibility to be part of international sarcoma trials. Therefore strong cooperation within the community of physicians treating sarcoma patients outside and within a sarcoma centre is needed.
Clinical history
Because an exact histologic classification of a soft tissue mass is essential for diagnosis and treatment planning, pathologists need to have an adequate clinical history at the time when tissue is submitted. Knowledge about patient’s sex and age, the size and the depth of the tumour mass (in relation to the superficial fascia, i.e., superficial above the fascia, deep in contact with the fascia or intramuscular), the relationship to adjacent structures (nerves, vessels, joints), the presence/pattern of calcifications, history of radiation, neurofibromatosis, hypoglycaemia in solitary fibrous tumour, or osteomalacia in phosphaturic mesenchymal tumour, speed of growth and pain pattern are helpful information for pathologists. In addition, providing imaging studies is always of diagnostic help.
To enable adequate histologic diagnosis on biopsy specimens multiple cores (with a 14–16 G needle) from different tumour areas need to be submitted to pathology ([2, 4]; Fig. 1).

Pathologic diagnosis is a step-by-step process starting with good clinical history and adequate tumour material. Gross examination (adequate sampling for histology), histology, immunohistochemistry and molecular pathology are reviewed by a specialized pathologist and included in the pathology report
A standard questionnaire/checklist concerning the clinical history, by including all the parameters already mentioned, would highly facilitate the diagnostic process.
Gross description
For pathologists it is extremely useful to examine the resection specimen together with the surgeon. Surgeons can highlight the critical margins and re-build the in vivo configuration of the specimen. If this optimal setting is not possible, the resection specimen must be labelled in a way that topographic orientation and exact evaluation of resection margins is possible. Standard gross description parameters include the following: (1) measuring the specimen in three dimensions, (2) giving the closest distance of each margin from the edge of the tumour in millimeter (mm) or centimeter (cm), (3) the tumour relationship with anatomical structures, (4) the depth (subcutis, fascial, intramuscular) of the tumour, (5) the size of the tumour, and (6) presence of necrosis, as percentage of the total tumour. Individual blocks need to be taken from the margins and generally one block per cm of the maximum diameter of the neoplasm needs to be taken for histologic evaluation [5,6,7]. A photo documentation of a representative cross section is always helpful (Figs. 1 and 3a, b).
Tumour classification
Pathologic diagnosis should be made according to the recent World Health Organization (WHO) classification “Tumours of Soft Tissue and Bone” (most recent 2013 edition—a new edition will be published soon, 2020) [3]. In addition, the mitotic rate (number of mitoses in the most proliferative area/10 high-power fields), the percentage of necrosis and for primary untreated tumour and grading should be provided. The Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) grading system is most commonly used [8]. Nevertheless there are tumours which are per definition low or high grade and the FNCLCC is not working.
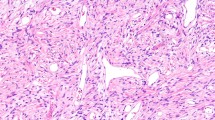
Traditionally STT have been classified based on the line of differentiation—meaning which normal cell type the neoplastic cells closely resemble. Such linage is assigned by the histologic appearance of the tumour as well as patterns of protein expression assessed by using a panel of immunohistochemical stains. Although most soft tissue tumours show a mesenchymal or neuroectodermal differentiation, a subset demonstrate unusual lines of differentiation not generally expected to be found in soft tissue (e.g. epithelial, myoepithelial, melanocytic). For other STT the line of differentiation, despite extensive IHC (immunohistochemistry), cannot be assessed leaving these tumours in the “undifferentiated” sarcoma category. Finally one must be aware that another subset of sarcomas can be classified based on their genetic fingerprint in combination with H&E morphology and IHC ([7]; Fig. 1).
Immunohistochemistry
Immunohistochemistry (IHC) plays a central role in the diagnosis of soft tissue tumours and should be routinely used. Traditional and widely available IHC stains are used to identify specific proteins in tumour cells indicating a specific line of differentiation (e.g. desmin and MyoFD5 or myoglobin expression in tumours with skeletal muscle differentiation). However, new molecular techniques enabled detection of novel highly specific markers for identifying several specific types of STT (e.g. MUC4 for low grade fibromyxoid sarcoma or STAT6 for solitary fibrous tumour). Other IHC stains for example panTRK (Fig. 2 left) or PDL1 are used to screen for treatment targets. The availability of a broad spectrum of IHC stains, the careful interpretation of these stains and the awareness of overlapping IHC in different STT is essential in soft tissue pathology (Figs. 1 and 3c).

Left: Nuclear and cytoplasmic expression with an antibody against panTRK (Ventana Assay). Right: NGS (Archer fusion plex sarcoma panel) revealed a NTRK3-ETV6 Fusion

a Radiologic image of a soft tissue mass (knee) with involvement of tendons. b Gross examination of a multinodular soft tissue tumour. c Histology and immunohistochemistry of a malignant soft tissue mass with expression of “melanocytic” markers. d Next generation sequencing demonstrated a EWSR1-ATF1 fusion confirming the diagnosis of clear cell sarcoma of soft parts
Genetic classification
Soft tissue tumours can be divided into a group of tumours with simple genetic changes (e.g. translocations/fusions in Ewing sarcoma, synovial sarcoma, low grade fibromyxoid sarcoma), or tumours with single-gene mutations (e.g. GIST or desmoid) where genetic fingerprint facilitate a specific tumour diagnosis or guide patient’s treatment (e.g. Kit mutations or NTRK1-3-fusions) (Fig. 2 right). The other group includes tumours with aneuploidy and complex cytogenetic features, where the genetic fingerprint (at the moment) can only be individually used. Especially in the first mentioned group molecular diagnostics enables us to confirm and to guide diagnoses by an “independent” method. Molecular fingerprints will be more and more included to guide tailored treatment for patients with STS. To offer patients these opportunities, tissue needs to be sent to specialized centres for evaluation ([9], Figs. 1 and 3d).
Sarcoma centres provide experience and expertise in sarcoma pathology
Expertise in sarcoma pathology is usually available in sarcoma centres providing the following: (1) A team of sarcoma specialists including surgical, orthopaedic, medical oncologists, radiologists, pathologists, oncology nursing as well as rehabilitation services; (2) Weekly established sarcoma conferences (e.g. tumour board); (c) Treating at least 100 sarcoma patients per year; (d) Availability of CT, MRI and PET scans; (e) Group members should belong to sarcoma-oriented medical organizations and publications concerning sarcomas in peer-reviewed journals should be presented by the group.
Several guidelines highlight the need for a second opinion in sarcoma diagnosis, if the diagnosis has been made outside a sarcoma centre [2]. Studies documented that a second opinion led to changes in diagnosis in up to 40% of cases with considerable impact on patients management or for example changes in tumour grading in 25%, or changes in the margin status from “negative” to “positive” in 49% of cases [10, 11]. This means that more than a third of diagnoses made by a general pathologist will be revised when the case is reviewed by a STS pathologist.
This is caused by three main reasons:
-
Pathologist experience is dependent on frequent exposure to soft tissue neoplasms—and these tumours are rare.
-
Laboratory expertise in managing sarcoma specimen—from gross sampling to availability of specific immunohistochemical or molecular assays is essential in sarcoma pathology.
-
Cooperation with a multidisciplinary sarcoma team is needed to diagnose and treat sarcoma patients.
The notion that sarcoma patients treated in high-volume centres have a significantly better survival and functional outcomes is demonstrated by a meticulous study of 4205 patients [12].
Take home message
Soft tissue sarcomas are rare cancers with more than 100 different subgroups and tremendous morphologic heterogeneity. Based on the fact that pathologic diagnosis dictates prognosis and treatment, patients suffering from this orphan disease should receive a second opinion from a soft tissue pathologist before a final treatment decision is made. Patients should be mandatorily treated in specialized centres by a multidisciplinary team in close cooperation with physicians interested in this orphan disease.
References
Stiller CA, Trama A, Serraino D, et al. Descriptive epidemiology of sarcomas in Europe: report from the RARECARE project. Eur J Cancer. 2013;49:684–95.
Casali PG, Abecassis N, Bauer S, et al. Soft tissue and visceral sarcomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Supplement 4):iv51–iv67.
Fletcher CDM, Bridge JA, Hogendoorn PC, Mertens F, editors. WHO Classification of Tumours of Soft Tissue and Bone. Lyon: IARC; 2013.
von Mehren M, Randall RL, Benjamin RS, et al. Soft Tissue Sarcoma, Version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16:536–63.
Fisher C. Standards and datasets for reporting cancers. Dataset for histopathological reporting of soft tissue sarcomas. Royal Coll Pathol. 2017; 1–24. https://www.rcpath.org/uploads/assets/369456b0-f7b4-4631-a0c6d161176f5782/Dataset-for-histopathological-reporting-of-soft-tissue-sarcoma.pdf. Accessed 5 Feb 2020.
Rubin BP, Cooper K, Fletcher CD, et al. Protocol for the examination of specimens from patients with tumors of soft tissue. Arch Pathol Lab Med. 2010;134:e31–e9.
Brodowicz T, Amann G, Leithner A, et al. Konsensus Diagnose und Therapie von Weichteilsarkomen. Wien Klin Wochenschr 2012;124(3–4):85–99. https://doi.org/10.1007/s00508-011-0079-8. Epub 2011 Oct 28.
Trojani M, Contesso G, Coindre JM, et al. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer. 1984;33:37–42.
Szurian K, Kashofer K, Liegl-Atzwanger B. Role of next-generation sequencing as a diagnostic tool for the evaluation of bone and soft-tissue. Tumors Pathobiol. 2017;84(6):323–38. Review.
Ray-Coquard I, Montesco MC, Coindre JM, et al. Sarcoma: concordance between initial diagnosis and centralized expert review in a population-based study within three European regions. Ann Oncol. 2012;23:2442–9.
Randall RL, Bruckner JD, Papenhausen MD, et al. Errors in diagnosis and margin determination of soft-tissue sarcomas initially treated at non-tertiary centers. Orthopedics. 2004;27:209–12.
Gutierrez JC, Perez EA, Moffat FL, et al. Should soft tissue sarcomas be treated at high-volume centers? An analysis of 4205 patients. Ann Surg. 2007;245:952–8.
Funding
Open access funding provided by Medical University of Graz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
B. Liegl-Atzwanger declares that she has no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liegl-Atzwanger, B. The role of pathology in sarcoma. memo 13, 159–163 (2020). https://doi.org/10.1007/s12254-020-00574-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12254-020-00574-4