
Abstract
Kaposi’s sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus-8 (HHV-8), is etiologically linked to the development of Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. These malignancies often occur in immunosuppressed individuals, making KSHV infection-associated diseases an increasing global health concern with persistence of the AIDS epidemic. KSHV exhibits biphasic life cycles between latent and lytic infection and extensive transcriptional and posttranscriptional regulation of gene expression. As a member of the herpesvirus family, KSHV has evolved many strategies to evade the host immune response, which help the virus establish a successful lifelong infection. In this review, we summarize the current research status on the biology of latent and lytic viral infection, the regulation of viral life cycles and the related pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus-8 (HHV-8), is the etiological agent of Kaposi’s sarcoma (KS) and is also linked to two other lymphoproliferative disorders, primary effusion lymphoma (PEL) and a plasmablastic form of multicentric Castleman’s disease (MCD) (Chang et al.1994; Dupin et al.1999; Soulier et al.1995). Recent studies have reported a disease called KSHV inflammatory cytokine syndrome, or KICS, characterized by higher levels of interleukin-6 and high viral loads in patients (Uldrick et al.2010).
KSHV is the γ2-herpesvirus, as it has a similar double-stranded linear DNA genome, but it varies in length from approximately 165–170 kb. The genome comprises a unique central coding region of approximately 145 kb and both sides of the region comprise highly GC-rich terminal repeats (TRs) (Russo et al.1996). KSHV has some highly conserved genes from open reading frame (ORF) 4 to ORF75 in a consecutive order. Similar to other herpesvirus members, these genes typically encode proteins associated with viral replication and structural virion components (Neipel et al.1997; Russo et al.1996). In addition, KSHV encodes a unique set of genes, designated with the prefix K (K1–K15), that have multiple functions in viral infection and virus-induced diseases (Ganem 1997; Russo et al.1996), a set of viral microRNAs (miRNAs), and several long non-coding RNAs including highly abundant lytic 1.1 kb polyadenylated nuclear RNA (PAN) (Staskus et al.1997). The basic structure of the KSHV virion is an electron-dense nucleocapsid surrounded by a lipid bilayer envelope. The layer between the envelope and capsid is called the tegument, which is divided into an outer and inner layer, although this division is not very clear. The envelope consists of seven glycoproteins, including ORF8 (glycoprotein B, gB), K8.1, ORF22 (gH), ORF47 (gL), ORF39 (gM), ORF53 (gN) and ORF68 (Akula et al.2001; Baghian et al.2000; Koyano et al.2003; Naranatt et al.2002; Wang et al.2001). The tegument proteins include ORFs 45, 52, 11, 21, 33, 63, 64 and 75, and are thought to possibly contribute to early viral replication events and genome entry upon primary infection (Bechtel et al.2005; Full et al.2014; Gillen et al.2015; Gregory et al.2011; Lan et al.2005b; Li et al.2016a; Lock et al.2002; Sathish et al.2012; Zhu et al.2005). In addition, several abundant host cytoplasmic proteins, such as HSP90, HSC70, EF-2B and actin are also found in the virions (Bechtel et al.2005) (Fig. 1A).

KSHV virion structure and clinical manifestations of Kaposi sarcoma. A Schematic presentation of KSHV structure. KSHV virions display the icosahedral nucleocapsids surrounded by a lipid bilayer envelope. Between the capsid and envelope is a morphologically amorphous layer called the tegument. The viral proteins found in KSHV capsid, tegument, and envelope are listed below. B Lesions of classical Kaposi’s sarcoma patients in Xinjiang province of China. The photos were provided by Dr. Tiejun Zhang from Fudan University School of Public Health.
Epidemiology of KSHV Infection
KS was first described as skin lesions, typically occurring among Mediterranean or Ashkenazi Jewish elderly men, by a Hungarian doctor named Moritz Kaposi in the late 19th century. Prior to the HIV epidemic, KS was found to be common only in certain geographical areas, such as the Mediterranean region and sub-Saharan Africa (Cook-Mozaffari et al.1998; Dollard et al.2010). However, with case reports of unusual KS occurring in young men having sex with other men (MSM) in the United States in the 1980s, KS became to be an AIDS-defining malignancy. In contrast to the speculation that HIV was the etiological agent of KS, Chang et al. first identified KSHV genomic DNA by performing representational differential analysis of KS lesions and normal tissue in 1994 (Chang et al.1994). To date, KSHV has been confirmed as the causative agent of KS.
According to reported clinical studies, KS is divided into four classes: classic KS, which develops especially in Jewish elderly men (Iscovich et al.1998); endemic KS, occurring in sub-Saharan Africa evenly in adults and children (Revilla-López et al.2015); AIDS-associated KS, closely associated with HIV infection; and iatrogenic KS, generally occurring in the recipients who receive the immunosuppressive therapy after organ transplantation.
Compared to other human herpesviruses, KSHV infection is not ubiquitous, and the incidence of KS is geographically limited from sub-Saharan Africa to Europe and the US. Numerous methods have been used to clearly illustrate data regarding the KSHV seroprevalence in large population-based studies (Gao et al.1996; Lennette et al.1996; Simpson et al.1996). Currently, KSHV seroprevalence varies among different populations and three major patterns are observable: (1) high-level endemic areas, mainly occurring in Africa with seropositivity rates greater than 50%; (2) intermediate-level endemic areas, mainly located in Mediterranean countries with seroprevalence rates between 10% and 25%; and (3) nonendemic areas, mainly referring to most parts of Europe, Asia and the US with no more than 10% seropositivity in the population. Additionally, the prevalence increases in gay men in Western countries and Amerindians are reported to be more susceptible to KSHV infection than other ethnic groups. In China, KSHV seropositivity varies among different regions and ethnicities, as the virus mainly infects individuals of Xinjiang province located in the northwest of China and adults of Kazak and Uyghur ethnicities, at seropositive rates ranging from 20% to 40%. Therefore, Xinjiang is considered an endemic KSHV region in China. Interestingly, KSHV has a higher seroprevalence in people of Han ethnicity in Xinjiang than in other Han populations in the rest of China, but the reason underlying the difference in the geographical and population-based distribution of KSHV incidence remains unknown (Cao et al.2014; Fu et al.2009; Liu et al.2017; Minhas and Wood 2014; Wang et al.2010; Zheng et al.2017) (Fig. 1B).
The modes of KSHV transmission vary among different regions and populations. In nonendemic regions, sexual contact is the main route of transmission, with evidence of an elevated prevalence in gay men (Martin et al.1998; Melbye et al.1998). However, there are no direct evidences that sexual contact among heterosexual individuals could possibly get the infection. Different studies are inconsistent about the transmission mode. Studies conducted among sexual workers in Africa have suggested that sexual transmission indeed occur in adults (Eltom et al.2002; Lavreys et al.2003), but another study conducted in South Africa showed no direct evidence of sexual transmission (Malope et al.2008). In addition, a study conducted among the Iranian general population and intravenous drug users (IVDUs) with high-risk sexual behaviors demonstrated that KSHV infection was more prevalent in the IVDUs compared with healthy subjects and the findings indicate the likelihood of sexual route of KSHV transmission in the Iranian population (Kakavand-Ghalehnoei et al.2016). Another study from China reported that KSHV prevalence is similar in HIV-negative heterosexuals, female sex-workers, and sexually transmitted infection (STI) male patients, but represents approximately 20% and 30% in IVDUs and MSM, respectively (Zhang et al.2017). The study also indicates that sexual transmission of the virus is important in MSM but not in heterosexuals (Zhang et al.2017).
Besides sexual transmission, peripheral blood analysis results suggest that KSHV can be transmitted via blood and blood products (Dollard et al.2005; Whitby et al.1995), although the viremia in KSHV infected patients was uncommon and the specific cell-associated nature didn’t support efficient transmission. However, studies conducted in Uganda, where HHV-8 is endemic, provided a strong evidence that KSHV is transmitted by blood transfusion (Hladik et al.2006). Further studies showed that transfusion of short stored blood presents increased risk of death (Hladik et al.2012). The high prevalence in children especially in endemic regions leads to the possibility of transmission from mother to child vertically and some reports support the hypothesis in part, but more samples and studies are needed to further exploration (Lisco et al.2006; Mantina et al.2001).
Apart from the routes discussed above, saliva exchange in a nonsexual manner may be an important mechanism of viral spread among children and adults in the endemic regions, as viral DNA is detectable in their saliva (Boldogh et al.1996; Martin et al.1998; Smith et al.1999).
Life Cycles of KSHV
KSHV infects multiple cell types including B lymphocytes, endothelial and epithelial cells, typically via membrane fusion mediated by several viral glycoproteins, such as gB, gH and gL (Avitabile et al.2009; Krishnan et al.2005; Pertel 2002; Veettil et al.2014). In addition, some host proteins, including heparan sulfate, integrin, ephrin A2, xCT and dendritic cell-specific ICAM-3 grabbing nonintegrin (DC-SIGN), can serve as receptors for KSHV and facilitate attachment of the virion to the cell membrane (Akula et al.2002; Hahn et al.2012; Hensler et al.2014; Kaleeba and Berger 2006; Rappocciolo et al.2006). KSHV primary infection outcome and gene expression after infection vary in different target cells, such as endothelial, epithelial, B cells, keratinocytes and monocytes, which predominantly establish persistent latent infection and express mainly the latent transcriptome (Jha et al.2014; Purushothaman et al.2015; Singh et al.2014). Specifically, infection of primary tonsillar lymphocytes with KSHV causes an abortive lytic infection of T cells (Myoung and Ganem 2011b). Therefore, latency is the default life cycle for KSHV following host cell infections (Giffin and Damania 2014).
Similar to other members of the herpesvirus family, KSHV displays two different modes of infection, latent infection and lytic reactivation, differentiated by complex but characteristic gene expression patterns (Cai et al.2010; Ye et al.2011a). During latency, the viral genome circularizes in the nucleus and maintains as a chromatinized nuclear plasmid; thus, no virion production occurs, and only a small subset of genes are expressed (Ballestas et al.1999). Latent DNA replication proceeds from multiple origins of replication (ori-P) in the TRs with the aid of host cellular DNA polymerase and a limited genome copy number is maintained via even distribution to daughter cells (Deng et al.2012; Lin et al.2003). The latent state is not invariable and can be disrupted by environmental and physiological stimuli, such as oxidative stress and hypoxia (Davis et al.2001; Ye et al.2011b). The switch may activate the resting genome, which features a substantial and sequential viral gene expression and viral DNA replication (Chen et al.2001; Haque et al.2003). During reactivation, lytic genes begin to be expressed in a temporally regulated transcriptional cascade as follows: (1) immediately early (IE) genes, which primarily encode transcription factors and regulators; (2) early genes, which function in preparation of the cell ready for viral DNA replication and viral protein production; and (3) late genes, which are subsequent to the onset of replication and comprise viral structural components (Carroll et al.2007; Davis et al.2015; Jenner et al.2001; Saveliev et al.2002; Zhu et al.1999). After all classes of the lytic genes are expressed, virus assembly begins in the nucleus. The replicated genomes are incorporated into newly synthesized capsids, acquire teguments, and finally bud through host membranes to obtain envelopes. Subsequently, viral progeny are released from the host cell (Gradoville et al.2000). The boundary between the latent and lytic state is not clear. In the KSHV lytic infection, virus-infected cells display both lytic and latent gene expression simultaneously. A brief description of KSHV life cycles is summarized in Fig. 2.

Diagram of KSHV life cycle events. Depicted are virion attachment, entry into the host cells and the different life cycles decision. KSHV entry is a multiple step involved in viral glycoproteins and many cellular membrane proteins. The entry is initiated by the binding of glycoproteins to specific cell receptors in the host cell membrane. After the entry, KSHV genome packaged in capsid is transported through the cytosol and then the genome is released into the nucleus. Once the viral genome enters the nucleus, the decision to enter the latent or lytic phase of the life cycle is made. KSHV life cycle contains two phases of infection: a short lytic replication and a persistent latent replication. During latency, LANA protein tethers KSHV episome to host cell chromosome and partitions certain numbers of genome copies to new cells. Upon exogenous stimuli, KSHV can be induced to lytic cycle. During lytic cycle, the KSHV genome replicates in a rolling cycle mechanism to produce linear genomes that are packaged into capsids. Mature capsids then obtain virus envelop by budding on host membranes. Lytic replication subsequently leads to destruction of the cells and release of virions to infect new cells.
Regulation of Viral Latency
Establishment of KSHV Latency
In terms of viral genome persistence in infected cells, KSHV has developed many mechanisms to establish long-term, nonproductive latent infection. To accomplish this goal after lytic infection, the viral genome has to first be circularized and chromatinized into a new form of epigenome to tightly control early gene expression in an off–on latent-specific pattern (Ballestas and Kaye 2001). Although the transcriptional pattern exhibits latency, the virus genome maintains the potential to enter the primary infection phase. Thus, suppression or silencing of viral lytic gene expression is the mechanism by which KSHV remains latent. Furthermore, during the centuries’ coevolution of the virus and host, KSHV has acquired a number of genes that selectively evade host immune system to maintain its persistent infection (Areste and Blackbourn 2009; Coscoy 2007; Lee et al.2010, 2012, 2016a).
Transcription Regulation of Latent Genes
Only a small portion of the latent KSHV genome is transcribed, and the major latency locus in latently infected PEL cells includes the genes for ORF73 (latency-associated nuclear antigen, LANA), ORF72 (v-Cyclin), ORF71 (K13) (Fas-associated death domain-like interleukin-1β-converting enzyme (FLICE)-inhibitory protein, v-FLIP), and K12/Kaposin (Kaposin A, B and C) and about 25 mature microRNAs (miRNAs) (Cai and Cullen 2006; Cai et al.2005; Dittmer et al.1998; Gottwein et al.2006, 2011; Kedes et al.1997; Staskus et al.1997). These genes, positioned in the minus strand of double-stranded viral genome, are transcribed from different latency promoters; LANA, v-Cyclin and v-FLIP are transcribed from the LANA promoter or LTc, while the others are transcribed from the Kaposin promoter, or LTd (Pearce et al.2005; Sadler et al.1999). In KSHV-infected MCD and PEL tissues, another latency locus encoding the vIRF3 gene was found to be expressed as the LANA-2 (K10.5), which is known to inhibit interferon induction and thus enhance cell proliferation and survival (Fakhari and Dittmer 2002). In addition, using array-based transcript profiling with limiting-dilution reverse transcription-PCR (RT-PCR), ORF K1 had been found to be weakly transcribed in most latently-infected cells, but remains upregulated during lytic reactivation (Chandriani and Ganem 2010). Similarly, examination of KS and PEL tumors also showed that several other viral genes including ORF74, K14, vIL-6 and ORF59, are transcribed at low levels (Majerciak et al.2013; Wen and Damania 2010), indicating a certain degree of the leaky expression of viral lytic genes during latent infection.
ORF73 (LANA)
LANA, the most abundant latent protein in both latently-infected cells and tumors, is about 1162 amino acids in length. The N-terminal region of LANA mainly associates with many chromatin-associated proteins, while the C-terminal region connects to numerous cellular proteins and chromosomes. The middle part of the protein contains a large repetitive region composed of acidic and glutamine-rich repeats resulting variable size of LANA protein (Ballestas and Kaye 2001; De Leon Vazquez and Kaye 2011; Kelley-Clarke et al.2009; Uppal et al.2014).
To maintain viral genome stability and replication, the C-terminus of LANA binds to the conserved TR repeats of the KSHV genome directly, while the N-terminal domain then loads the viral genome on host genome via interacting with chromatin proteins, thus avoiding genome loss during cell mitosis and maintaining a relatively low number of viral DNA copies in infected cells. Several cellular proteins have been reported to interact with LANA, including ATF4/CREB2/STAT3 for transcription, p53/pRb/von Hippel Lindau (VHL) for tumor repression, HP1/H2A/H2B/MeCP2/BRD4 for chromatin binding, and GSK-3β for signal transduction (Cai et al.2006; Fujimuro et al.2005; Garber et al.2001; Griffiths and Whitehouse 2007; Jr et al. 1999; Lim et al.2000; Ottinger et al.2006; Radkov et al.2000). LANA also directly binds to many epigenetic regulatory proteins, including DEK, nucleophosmin (NPM), CENP-F, DNMT3a, TRF1, topoisomerase II β (TopoII β), TIP60 and KDM3A (Krithivas et al.2002; Ottinger et al.2006; Purushothaman et al.2012; Sarek et al.2010; Shamay et al.2012; Xiao et al.2010). LANA function can also be regulated by post-translationally modifications of the protein. For example, PRMT1 mediates LANA lysine acetylation and arginine methylation, while Pim-1, CK1, GSK1, GSK3 and RSK3 lead to LANA phosphorylation (Bajaj et al.2006; Campbell et al.2012; Cheng et al.2009; Lu et al.2006; Woodard et al.2012). As reported, LANA binds to several viral lytic promoters to inhibit viral lytic gene transcription, which is an important mechanism for the maintenance of latency (Hu et al.2014). For example, LANA prevents RTA-mediated activation of lytic replication by competitive interaction with RBP-Jκ (Lan et al.2004, 2005a).
ORF72 (v-Cyclin)
ORF72 is the homolog of cellular cyclin D2 and similarly functions in regulating the cell cycle and cell proliferation by constitutive activation of cellular cyclin-dependent kinase 6 (CDK6) (Jones et al.2014; Li et al.1997; Van Dross et al.2005). The vCyclin-CDK6 complex was found to have a large range of substrates and phosphorylates cellular p21 and p27, resulting in accelerated G1/S phase transition in PEL cell lines (Chang and Li 2008; Godden-Kent et al.1997; Jarviluoma et al.2004; Laman et al.2001). In addition, nucleophosmin (NPM) was found to be phosphorylated by the complex to control latency program in KSHV-infected cells (Sarek et al.2010). Deletion of v-Cyclin with the homologous recombination method was found to affect cell proliferation and cell cycle progression in a density-dependent manner (Jones et al.2014).
ORF71 (K13, v-FLIP)
ORF71 or v-FLIP is a homolog of cellular FLICE inhibitory protein (Krueger et al.2001; Li et al.2006). The best characterized function of ORF71 is to activate a key cellular signaling pathway, NF-κB pathway, by directly binding to the IκB kinase γ (IKKγ) complex and thus facilitate cell survival, proliferation and cell type-specific induced growth arrest and apoptosis during latency (Bagneris et al.2008; Chaudhary et al.1999; Field et al.2003; Graham et al.2013; Grossmann et al.2006; Matta et al.2007). Furthermore, v-FLIP suppresses the AP-1 pathway by activating NF-κB pathway to inhibit viral lytic replication and promote latency (Ye et al.2008). In addition, many cellular proteins involved in the activation of NF-κB pathway are induced by v-FLIP via protein–protein interaction, such as cell adhesion molecule 1 (CADM1) and NEMO (Hunte et al.2018; Matta et al.2012). Recently, the v-FLIP coding sequence-deleted virus has been constructed to investigate more function of this gene in KSHV life cycles (Wang et al.2018).
Kaposins (K12)
In contrast to the three latent genes described above, the Kaposin locus is located downstream of the LANA promoter and encodes a complex of proteins, including Kaposins A, B and C (Sadler et al.1999; Sarid et al.1999). Kaposin A, a hydrophobic polypeptide, is mainly found on intracellular and cell surface membranes (Tomkowicz et al.2002). As the smallest isoform from the K12 locus, Kaposin A can induce focal transformation in vitro in Rat-3 fibroblasts and nude mice, indicating its oncogenic potentials in induction of the transformation phenotype (Muralidhar et al.1998). Kaposin A interacts with the guanine nucleotide exchange factor cytohesin-1 and activates ERK2/MAPK (Kliche et al.2001), while Kaposin B enhances the expression of cytokines by inhibiting the degradation of their mRNAs containing AU-rich elements (AREs) in their 3′ noncoding regions via binding to and activating p38/MK2 pathway (McCormick and Ganem 2005). In addition, Kaposin B was recently found to regulate microRNAs cooperated with c-Myc in KSHV-infected cells (Chang et al.2016; Corcoran and McCormick 2015). To date, no published studies have examined Kaposin C (Speck and Ganem 2010).
Viral miRNAs
miRNAs, a group of noncoding single-stranded RNAs approximately 19–23 nucleotides (nt) in length, primarily regulate gene expression at the posttranscriptional level by binding to the seed-matched regions of target mRNAs (Ameres and Zamore 2013). KSHV has been identified to encode 25 mature viral miRNAs from 12 viral pre-miRNAs (Cai et al.2005; Grundhoff et al.2006; Pfeffer et al.2005). Mature miRNAs are abundantly expressed in latently infected cells and tissues from patients with KS or MCD (Grundhoff et al.2006; Sullivan 2007), and according to published studies, some specific KSHV miRNAs were regulated at different levels in different phases of the viral life cycle (Qin et al.2017). Some miRNAs were also detected in KSHV virions and function in cellular communication, while a few miRNAs from patient exosomes were found to enhance cell migration (Chugh et al.2013; Lin et al.2012).
As reported, KSHV miRNAs function in regulating virus life cycles, cell immune response, virus-induced angiogenesis and spreading of KS by targeting and inhibiting the expression of multiple cellular and viral genes. For instance, miR-K9-5p and miR-K12-7 together with miR-K12-9 were identified to inhibit RTA expression, resulting in maintenance of viral latency (Bellare and Ganem 2009; Lin et al.2011). Other miRNAs like miR-K11 and miR-K3, also target cellular factors, such as IκB α, BCLAF1 and nuclear factor (I/B) to promote latency (Lei et al.2010; Lu et al.2010; Ziegelbauer et al.2009). A role of miR-K3 in maintaining viral latency is to directly target G protein-coupled receptor kinase 2 (GRK2) to activate CXCR2/AKT signaling and promote endothelial cell migration and invasion (Hu et al.2015; Li et al.2016b). In addition, at least two KSHV miRNAs were found to functionally mimic cellular miRNAs by targeting same transcripts, including miR-K12-11 mimicking hsa-miR-155 (Gottwein et al.2007; Skalsky et al.2007) and miR-K10a mimicking hsa-miR-142 (Forte et al.2015). KSHV miRNAs also manipulate host immune surveillance to promote latency. For instance, miR-K12-11 controlled the IFN signaling by targeting IKKε to suppress antiviral immunity, resulting in establishment of long-term latency (Liang et al.2011). miR-K12-7 targets the stress-induced immune molecule (MICB) during virus infection to avoid the attacks by natural killer (NK) cells (Nachmani et al.2009). miR-K10a targets tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) and the inhibition prevents TWEAK-induced apoptosis and inflammatory cytokine (IL8) expression (Abend et al.2010). miR-K9 and miR-K5 target the 3′UTR of interleukin-1 receptor (IL-1R)-associated kinase (IRAK1) and myeloid differentiation primary response protein 88 (MYD88) respectively, the overexpression of the miRNAs leads to the reduction of secreted IL-6 and IL8 in HUVEC cell culture (Abend et al.2012).
KSHV miRNAs also play important roles in KS angiogenesis and development. miR-K2 and miR-K5 target and reduce the different isoforms of tumor suppressor protein tropomyosin 1 (TPM1), which both lead to enhanced tube formation and VEGFA expression critical to KSHV angiogenesis and pathogenesis (Kieffer-Kwon et al.2015). Breakpoint cluster region mRNA (BCR) was also identified as a target of miR-K6-5, and the suppression increased the tube formation in HUVECs transfected with miR-K6-5 in vitro and lytic reactivation in BCBL-1 cells (Ramalingam et al.2015). miR-K6-3p was found to promote endothelial cell migration and angiogenesis by targeting 3′ UTR of SH3 domain binding glutamate-rich protein (SH3BGR) and activating STAT3 pathway (Li et al.2016c). Recently, miR-K6-5p was found to target metastasis suppressor CD82 while inhibition increased the cell invasion and angiogenesis by activating c-Met signaling (Li et al.2017).
Suppression of Viral Lytic Genes
Broad repression of viral lytic gene expression is one of the mechanisms by which latency is established and maintained. After de novo infection, the viral genome has to be first chromatinized, and the viral LANA protein then recruits the host polycomb repressive complexes (PRC1 and PRC2) to the promoters of lytic genes to initiate latency establishment (Toth et al.2013b, 2016). Depletion of the LANA protein in the context of the KSHV genome decreased the recruitment of PRCs to the viral genome during infection. Therefore, LANA can function as a genome-wide repressor of lytic gene expression. Studies have also reported that LANA directly binds to and inhibits RTA promoter activities. Moreover, LANA indirectly and directly binds to other transcription activators and repressors, such as Sp1, RBP-Jκ and KAP1, all of which can regulate the expression of RTA (Lan et al.2004, 2005a; Sun et al.2014b; Verma et al.2004). Experiments performed using small interfering RNA in PEL cells or using the LANA deletion mutant BAC36ΔLANA showed that LANA could repress the viral lytic reactivation phase, as it repressed the expression of all classes of viral lytic genes and the final production of infectious KSHV virions (Li et al.2008).
Epigenetic Regulation of Latency
Like other herpesvirus members, KSHV latency is governed in part by epigenetic modifications. To maintain the quiescent state of limited latent gene expression, some critical lytic genes are silenced, but treatment of latently infected PEL cells with DNA methyltransferases and histone deacetylases inhibitors induces lytic reactivation and replication (Miller et al.2007). Epigenetic modifications of the herpesvirus genome are mainly characterized as three groups: DNA methylations, chromatin protein posttranslational modifications and higher-order chromosome conformations (Chen et al.2013).
DNA Methylation
DNA methylation patterns are important for regulating KSHV latency (Gunther and Grundhoff 2010), and DNA methylation is typically indicative of repressed transcription and stabilized viral latency. To explore the potential roles of such modifications of KSHV gene expression, initial studies focused on the promoters of ORF50 and ORF73/LANA, two key molecules during the KSHV life cycle. However, subsequent studies on KSHV-positive tumor samples did not yield similar results, as the RTA promoter was heavily methylated in BCBL-1 cells, while the LANA promoter was not. Furthermore, the global methylation status of viral episomes has been examined in PEL derived cell lines using MeDIP (methylated DNA immunoprecipitation) technology, revealing that DNA in the KSHV genomes is profoundly methylated during latency (Gunther and Grundhoff 2010). The genome mainly includes regions upstream of the LANA promoter (LANAp) and other locations, including K7, K9, ORF45/50 and ORF8 (Gunther and Grundhoff 2010).
Histone Modifications
Histone modifications have also proven to be indispensable for regulating KSHV latency. These markers include the active histone acetylation markers histone H3 and H4N-terminal tails (e.g. H3K9/K14-ac) and trimethylation of H3 at lysine 4 (H3K4-me3) (Lu et al.2003) as well as, the repressive histone modifications of lysine 9 trimethylation in histone H3 (H3K9-me3), a hallmark of constitutive heterochromatin, and trimethylation of lysine 27 (H3K27-me3).
High-resolution genome-wide studies on histone modifications during latency and reactivation have been conducted, revealing a distinctive pattern of activating and repressing markers during latency and inducing changes upon reactivation. The latency-associated locus was shown being enriched by activating the H3K4-me3 and AcH3 histone modifications, but no repressive H3K9-me3 and H3K27-me3 markers. DZNep, a small molecule inhibitor of the H3K27-me3 methylase, was shown to stimulate KSHV lytic cycle gene activation, suggesting a positive role of H3K27-me3 in latency establishment and maintenance (Bernstein et al.2006). The promoter regions of RTA and other early genes are associated with both activating (H3K4-me3) and repressing (H3K27-me3) histone modifications, which indicates multiple and dynamic mechanisms of regulating chromatin structure during viral gene expression (Toth et al.2010, 2013a). In terms of latency maintenance, histone modifications function similarly to the on–off switch of the latent state and reactivation phase by selectively expressing latent and lytic loci in the KSHV genome (Toth et al.2013b).
As noted above, histone modifications have profound impacts on the existence of the virus in cells, and the factors responsible for managing histone modifications and nucleosome positioning may thus be substantially relevant for understanding the epigenetic control of KSHV latency. CCCTC-binding factor (CTCF) is known to function as a chromatin-organizing factor (Dorsett 2011; Herold et al.2012; Ohlsson et al.2001; Van Bortle and Corces 2013a). However, a study on the KSHV genome-wide histone modification following de novo infection of SLK cells indicated an increased H3K27-me3 for K12 but no change for LANA, whereas the viral genome exhibited a remarkable increase of H3K4-me3 for both K12 and LANA in the course (4–72 h) of KSHV infection. Unfortunately, the heterochromatin hallmark H3K9-me3 on these protected sites, such as the KSHV LANA promoter region was not included in this study (Toth et al.2013b), raising the question about their biological significance of the reported histone modifications in the lytic-to-latent switch. Whether the chromatin boundary element prevents the protected regions from being occupied by repressive markers needs to be uncovered. The impaired episome stability after deletion of CTCF from the LANA promoter region confirmed this speculation (Chen et al.2012), but whether this loss is associated with an increase in H3K9-me3 or DNA methylation need to be further explored.
Chromatin Conformation
Higher-order chromatin structures, such as those resulting from promoter-enhancer DNA loop interactions, have been found on KSHV chromatin to influence the coordinated control of gene expression (Kang H et al. 2011; Van Bortle and Corces 2013b). A DNA loop with CTCF-cohesin binding sites in the LANA promoter region was found to interact with the ORF50 promoter region and regulate the lytic gene expression (Kang H et al. 2011). Disruption of the DNA loop stability, which mainly depends on cellular factors involved in sister-chromatid cohesion, led to reactivation of the lytic cycle, suggesting the importance of this chromatin conformation in maintaining latency (Chen et al.2012).
Mechanism of KSHV Latent Replication
To maintain the potential of the silenced virus genome during latency, KSHV has evolved its own strategy to simultaneously replicate the viral genome and cellular DNA. Furthermore, the replicated genome has to be precisely passed to daughter cells and keep effectively maintained at 10–150 genome copies per infected cell (Li et al.1996; Renne et al.1996a; Sturzl et al.1997; Wen and Damania 2010).
DNA replication is a multistep and sequential event that begins with the assembly of pre-replication complex (pre-RC) proteins at the replication origin. Generally, DNA replication is initiated at many replication origins almost simultaneously such that the process is completed within a limited time frame. Replication of the KSHV viral genome has been shown to initiate at multiple points associated with the TR region that contains an origin of replication, ori-P (Verma et al.2011). LANA can directly bind to ori-P via LANA-binding sites (LBS-1/2) and a 32-bp GC-rich segment (Hu and Renne 2005; Ohsaki and Ueda 2012; Verma et al.2007a), thus triggering the recruitment of various (pre-RC) proteins to ori-P. The pre-RC contains at least the origin recognition complex (ORC), poly(ADP-ribose) polymerase 1 (PARP1) and minichromosome maintenance proteins (MCM) within its nuclear matrix region (Lim et al.2002; Ohsaki et al.2004; Stedman et al.2004). Interestingly, LANA also interacts with and recruits cellular machinery to support latent replication. The cellular machinery recruited by LANA includes bromodomain containing 2 (BRD2), origin recognition complex 2 (ORC2), H4-specific histone acetylase (HBO1) and CREB-binding protein (CBP) (Stedman et al.2004), and the interactions occur in the carboxy-terminus of LANA, i.e., the TR binding domain (DBD). Subsequent to the recruitment of ORCs to the replication origin, some DNA replication licensing factors, such as MCMs, are directed to the same sites for replication (Tsuyama et al.2005).
LANA also interacts with replication factor C (RFC) ATPase, resulting in the augmentation of PCNA loading onto the DNA to facilitate the replication and persistence of viral DNA (Sun et al.2014a). PCNA is identified as a DNA sliding clamp that increases the proximity of replicative DNA polymerase for better processivity and is an essential component of the DNA replication machinery (Moldovan et al.2007). In addition, BUB1, a cellular mitotic kinase, was found to enhance the monoubiquitination of PCNA in the presence of LANA, which contributes to episome replication and maintenance (Sun et al.2015). Further studies have shown that LANA also interacts with and recruits cellular DNA topoisomerase 2-β (TopoII β) to the TR region. The main function of TopoII β protein is regulating and modifying the DNA topology by introducing double-stranded breaks, and thus plays a critical role in viral DNA replication (Purushothaman et al.2012).
In addition to ori-P, ori-A has been identified as another initiation site of latent DNA replication that is independent of LANA (Verma et al.2007b, 2011). Single molecule analysis of replicated DNA (SMARD) of the entire KSHV genome verified that more than one site in the genome can initiate replication. Chromatin immunoprecipitation (ChIP) experiments to detect ORC2 and MCM3 accumulation suggested that various regions of the genome might potentially serve as the replication sites (Verma et al.2011).
Lytic Reactivation
The latent state of KSHV infection is reversible and inducible. Once the latency is disrupted by physiological factors or an exogenous stimulus, the virus begins the new phase of its life, lytic reactivation, by consecutively expressing most viral genes, i.e., immediate early, early and late genes, finally resulting in the production of infectious virion particles.
As a substantial number of viral genes are expressed, viral DNA is amplified, and the virus simultaneously exploits the cellular machinery to generate new infectious virions that are released from the infected cells to spread the virions to other susceptible cells. To better promote survival and reproduction, KSHV does not kill the resident host. Furthermore, as latent viral infection is the outcome occurring most often, the biological significance of the virus having both a latent and a lytic cycle are obvious: latent phase for maintaining the silenced virus genome for long-term, but producing no viruses, but lytic phase for virus propagation and spreading the virus to a new host. The lytic cycle may be also important for virus-mediated disease development. The viral load in peripheral blood mononuclear cells in patients correlates with KS progression (Quinlivan et al.2002). To control the KSHV-mediated malignancies in KS patients and AIDS individuals who are susceptible to KS, some drugs that block viral lytic DNA replication, such as foscarnet and ganciclovir, have been proven to repress early KS. In most KS lesions, the tumor-forming spindle cells are latently infected, but a small percentage of cells still undergo spontaneous lytic replication. Collectively, the lytic cycle not only plays an important role in the intact virus life cycle for virus propagation, but also has indispensable functions in KSHV pathogenicity.
Various host and experimental factors trigger virus reactivation from latently infected cells. To investigate the mechanism underlying the switch between the latent and reactivation states, several chemical reagents, including 12-O-tetradecanoyl-phorbol-13-acetate (TPA) (Miller et al.1997), sodium butyrate, valproic acid, ionomycin, and 5-azacytidine, have been widely used (Al-Kzayer et al.2016; Lukac et al.1999b; Shin et al.2014). While KSHV infection is necessary for the development of KSHV-associated diseases, it is not sufficient to induce tumorigenesis (Andrei and Snoeck 2015; Ganem 2010). Several infectious pathogens known to occur simultaneously with KSHV-associated oncogenesis include HIV, Epstein-Barr virus (EBV), human cytomegalovirus (HCMV), human herpesvirus-6 and -7 (HHV-6 & -7), herpes simplex virus type 1 and type 2 (HSV-1 & -2) and human papillomaviruses (Adams et al.1995; Tang et al.2012). Reports conducted in PEL cells have proven that hypoxia may contribute to KSHV lytic reactivation (Davis et al.2001). In addition, oxidative inhibition can be a strategy for inhibiting viral replication and tumorigenesis, as treatment with antioxidant/H2O2 scavengers inhibits KSHV lytic replication and tumor progression in vivo (Ye et al.2011b). After initiation of the virus lytic cycle, many viral proteins and cellular factors are involved in regulation of the lytic events.
Viral Proteins with Transactivation Functions
ORF50 (Replication and Transcription Activator, RTA)
RTA plays a major role in KSHV lytic reactivation from viral latency. RTA mRNA is about 3.4 kb in length and has seven spliced isoforms. Because RTA utilizes a poly A signal downstream of K8.1 for its RNA polyadenylation, its 3′UTR region overlaps with K8 and K8.1 region (Tang and Zheng 2002; Zheng 2003). RTA can be detected within 4 h of primary infection (Purushothaman et al.2015) and in TPA-induced BCBL-1 (PEL) cells (Dourmishev et al.2003; Sun et al.1999). As a transcription factor, RTA is localized in the nucleus and contains two nuclear localization signals (NLSs).
RTA itself is essential and sufficient for KSHV reactivation (Lukac et al.1999a). KSHV genome with RTA deletion is defective in expression of early lytic viral genes, virus production and DNA synthesis. Many studies have demonstrated the RTA transactivates a series of important lytic genes, including viral early genes (such as PAN, thymidine kinase [TK or ORF21], vIL-6 [K2], ORF57, ORF59, K8, K9, K1, DBP [ORF6], and DNA polymerase [ORF9]) and the late genes, assembly protein (AP) and gB (ORF8) (Byun et al.2002; Chen et al.2009). RTA also transactivates the promoters of ORF52, ORF65, ORF56, ORF37 and vOX (K14) (Bu et al.2008; Chen et al.2009), and autoactivates its own promoter (Deng et al.2000). RTA transactivates viral promoters by two mechanisms: by directly binding to the RTA response elements (RREs) in the promoter, and by indirectly binding to the RREs with other cellular or viral proteins, such as the Notch signaling pathway effector recombination signal binding protein (RBP-Jκ) (Persson and Wilson 2010). Other cellular transcription factors involved in RTA transactivation activity are C/EBPα (CAAT/enhancer binding protein alpha), Oct-1 (octamer-binding protein-1), Sp-1, and XBP-1 (Carroll et al.2007; Lai et al.2011; Wang et al.2003; Wen et al.2009).
In addition to its transactivation ability, RTA also exhibits an ubiquitin E3 ligase activity, allowing cellular and viral proteins to undergo proteasome-mediated degradation. MYD88, a key adaptor of innate immunity to microbial infection, interacts with RTA in vitro and in vivo, and is a target of RTA E3 ligase activity, thus helping the virus evade innate immunity (Zhao et al.2015). RTA can be phosphorylated, ADP-ribosylated, ubiquitinated and modified into other forms (Campbell and Izumiya 2012; Ko et al.2012; Tsai et al.2012).
Apart from transcriptional regulation and proteasome-mediated degradation, RTA also activates lytic DNA replication by binding to C/EBPα and the RRE-binding motifs within the origin of lytic replication (ori-Lyt) for transcriptional activation and recruitment of additional factors.
K8 (Lytic Replication-Associated Protein)
The K8 encodes a protein designated as K-bZIP (K8α), belonging to the basic-leucine zipper (bZIP) family of transcription factors. The K-bZIP gene locus contains two promoters, one for early transcribed product K-bZIP, and one for late transcribed product K8.1 (Tang and Zheng, 2002). K-bZIP was discovered to interact with RTA and, in turn, repress the transactivation activity of RTA, which seems to play a negative role in viral lytic gene expression and replication (Liao et al.2003). K-bZIP induces p53 and p21 expression, directly interacts with CDK2 and promotes cell cycle arrest (Izumiya et al.2003). However, the function of K-bZIP could be antagonized by K8β, a truncated K-bZIP translated from a spliced K8 mRNA isoform (Yamanegi et al.2005). A recent study also showed that K-bZIP is not essential for lytic reactivation in KSHV BACmid systems, but is important for virus production in infected PEL cells (Wang et al.2011).
K-bZIP is associated with ori-Lyt-dependent DNA replication via its interaction with ori-Lyt (Lin et al.2003). In addition, K-bZIP interacts with HDAC to inhibit the recruitment of HDAC to the promoters of ori-Lyt and ORF50, thus repressing the RTA autoactivation (Martinez and Tang 2012). K-bZIP mediated transcription repression is, in part, associated with sumoylation at lysine residue 158. A genome-wide analysis showed that K-bZIP activated 21 promoters, while RTA activated 34 viral promoters (Ellison et al.2009). When combined, K-bZIP appears to repress three RTA-responsive promoters, suggesting additional role of K-bZIP in transactivating viral promoters during reactivation (Liao et al.2003). Collectively, K-bZIP has dual functions in inhibiting lytic gene expression under certain conditions and regulating the KSHV life cycle by promoting lytic DNA replication. Although not binding DNA, K8 binds T1.4 RNA to recruit viral DNA replication factors to ori-Lyt DNA and regulates KSHV DNA replication (Liu et al.2018).
Cellular Factors
Several cellular signaling pathways are involved in KSHV reactivation in latently infected cells, including PKC δ, PKA, b-Raf/MEK, p38 and JNK, Notch and RBP-Jκ, PI3K and Akt, TLR7/8 signaling and Pim-1/3 (Ford et al.2006; Gregory et al.2009; Haas et al.2013; Lan et al.2006; Xie et al.2005; Yu et al.2007). Protein kinase C δ plays a role in KSHV lytic replication, as overexpression of the dominant-negative PKC δ mutant supports an essential role for the PKC δ isoform in virus reactivation. Using specific pathway-targeted inhibitors, studies have found that b-Raf/MEK/ERK signaling plays a role in TPA-induced reactivation from latency. The JNK and p38/MAPK pathways were constitutively activated in latent BCBL-1 cells, but TPA treatment increased the levels of only activated ERK and p38 and reduced the expression of viral lytic genes, including RTA. Activation of MAPK pathways was shown to be necessary for activating the RTA promoter. After screening the human protein kinase cDNA library and analysis of complex cross-talk between cellular kinase pathways, Pim-1 and Pim-3 were identified as novel kinases involved in KSHV reactivation. Phosphorylation of LANA by Pim-1 and Pim-3 counteracts the LANA-dependent repression of viral transcription (Bajaj et al.2006; Cheng et al.2009). Similarly, screening involving TLR signaling in reactivation showed that TLR7/8 agonists reactivated latent KSHV and induced viral lytic gene transcription and replication. Apart from the above signaling pathways, ionomycin, a Ca2+ ionophore, was shown to induce RTA protein expression, which suggests a role of calcium-dependent signaling pathways in virus reactivation (Zoetweij et al.2000) (Fig. 3).

Regulation of KSHV latency and lytic reactivation. The purple are viral latent proteins, the brown are viral critical lytic proteins, the black is a few of cellular factors and signaling pathways involved in viral lytic cycle.
Posttranscriptional Regulation of Viral Gene Expression
Beside the regulation of transcription, the eukaryotic cells developed several additional mechanisms to regulate gene expression after transcription. As these processes occur after transcription, they are commonly referred as posttranscriptional and involve many cellular RNA-binding proteins and small RNAs. The first layer of such regulation comprises of proper processing of nascent transcripts including RNA capping, splicing, and polyadenylation to produce mature mRNA. Nuclear export of the mature mRNA to the cytoplasm and mRNA translation and turnover are the additional layers of posttranscriptional regulation of eukaryotic gene expression (Schaefke et al.2018). Recently, several groups found the newly transcribed viral RNA also harbors post-transcriptional N6-adenosine methylation marks and this event plays important roles in viral lytic replication based on the observation that some m6A sites are responsible for RTA pre-mRNA splicing and RTA increases its own expression through posttranscriptional mechanism (Ye 2017; Ye et al.2017). Further, the m6A epitranscriptome of KSHV had been mapped in diverse cell types both in latently infected cells and in cells undergoing lytic replication and the results showed that KSHV transcripts harbor wide-spread m6A modifications in both latent and lytic replication, of which YTHDF2, the ‘reader’ protein, responsible for multiple m6A function was found to inhibit lytic replication by enhancing stability of KSHV transcripts (Tan and Gao 2018; Tan et al.2017; Ye et al.2017).
Posttranscriptional Regulation of Latent RNA Transcripts
KSHV major latent locus is positioned at the minus strand of the double-stranded KSHV genome and consists of ORF73 (LANA)/ORF72 (v-Cyclin)/K13 (v-FLIP) and K12 (Kaposin). Posttranscriptional processing of the latent transcripts represents one of the best examples of such regulation during latent phase of KSHV infection. The latent locus expression is initiated from three alternative promoters, LTc and LTi upstream of ORF73 and LTd upstream of ORF72. Both LTc and LTd are constitutive promoters, whereas LTi is an RTA-inducible promoter (Pearce et al.2005). The primary RNA transcripts initiated from any one of the three latent promoters are tricistronic and polyadenylated either at a poly A site at nt 122,069 downstream of K13 or at nt 117430 downstream of K12 (Majerciak et al.2013). In addition to be regulated by alternative polyadenylation, the LTc trisitronic transcript also contains two introns with the intron 1 bearing two alternative 3′ splice sites for alternative RNA splicing to efficiently and selectively express the ORF72 and K13 ORF. The LTi tricistronic transcript has no intron and may be responsible for LANA expression in the lytic phase. The LTd tricistronic transcript has only one intron and encodes ORF72 and K13 if the intron escapes from RNA splicing, and otherwise, encodes K12 and KSHV viral miRNAs (Ajiro and Zheng 2014). Consequently, these alternative promoter usage, alternative RNA splicing and alternative polyadenylation lead to produce an array of latent mRNA transcripts with different coding potentials for expression of individual ORFs. Although almost all latent mRNA transcripts use cap-dependent translation, K13 may use an internal ribosome–binding site (IRES) for its translation (Bieleski and Talbot 2001). Because splicing of KSHV latent transcripts is required for the expression of KSHV-encoded miRNAs from the intron region, the excised intron spanning nt123595-118799 region must be processed by cellular miRNA processors to produce pre-miRNAs in the nucleus and mature viral miRNA in the cytoplasm (Cai and Cullen 2006; Pearce et al.2005). Unfortunately, the regulation of posttranscriptional processing of KSHV latent RNA transcripts remains poorly understood to date. One assumption is that there is no viral protein being involved in the posttranscriptional regulation of the latent transcripts.
Posttranscriptional Regulation of Lytic Gene Transcripts
KSHV genome encodes up to 80 lytic genes for its productive infection, of which more than one-third are split genes with introns and ~65 genes share a single or alternative pA sites with other genes for their RNA polyadenylation (Majerciak et al.2013). Thus, expression of these lytic genes is regulated by RNA splicing and polyadenylation in addition to transcription regulation. One of the well-studied lytic gene loci is the gene cluster region of RTA (ORF50), K8 and K8.1. All three genes are collineated side-by-side in the plus strand of the double-stranded viral genome and have their own promoters and gene-body introns, but share a single pA site downstream of K8.1. The expression of these three genes are in a cascaded order from immediately early (RTA), early (K8) to late (K8.1) stage of productive infection, but all utilize the same pA site for RNA polyadenylation. Thus, the transcripts from this region are polycistronic for RTA, bicistronic for K8 and monocistronic for K8.1 and subject to multiple levels of posttranscriptional regulation (Tang and Zheng 2002; Zheng 2003).
During lytic KSHV infection, viral genes are efficiently expressed to the levels required for virus replication and production. As described above, KSHV encodes RTA protein as a powerful transactivator to initiate transcription of most, if not all, viral lytic genes by binding to the promoter of individual genes. More importantly, KSHV encodes ORF57 (MTA, mRNA transcripts accumulation) as a powerful posttranscriptional regulator (Kieshner et al.2000; Majerciak and Zheng 2009) to secure that viral RNA transcripts could be processed properly and become stable, exportable from the nucleus to the cytoplasm and translatable in the cytoplasm. Thus, both RTA and ORF57 are equally essential for the robust expression of KSHV genes. In the absence of ORF57, most viral transcripts derived from RTA-mediated transcription are unstable and in short half-life because of lacking proper RNA processing (Han and Swaminathan 2006; Majerciak et al.2007, 2015).
ORF57 Expression and Protein Structure
Expression of ORF57 is activated in the early stage of KSHV infection by RTA binding to the ORF57 promoter (Duan et al.2001; Lukac et al.2001; Majerciak and Zheng 2009). The primary ORF57 transcript contains a small, constitutive intron in its 5′ half and a large, suboptimal intron in its 3′ half for alternative RNA splicing and is polyadenylated at a pA site 92 nts downstream of its coding region. This pA site is also used for expression of ORF56 upstream of ORF57 (Majerciak et al.2006b). The full-length ORF57 mRNA is derived from RNA splicing of the constitutive small intron, but retains the suboptimal large intron as the coding region. During KSHV lytic infection, only a small fraction (< 5%) of ORF57 RNA transcripts undergo double RNA splicing to produce a smaller, noncoding RNA isoform (Majerciak and Zheng 2016). ORF57 composes 455 amino acid residues and exhibits limited sequence homology to its homologues in other herpesviruses (Majerciak and Zheng 2009), but not at all to any known host proteins. Recent biochemical and crystallography studies revealed that ORF57 protein consists of two structurally and functionally distinct domains (Majerciak et al.2015; Yuan et al.2018) (Fig. 4). The N-terminal domain (NTD) encompasses first 219 amino acid residues and exhibits properties of an intrinsically disordered region (IDR), the naturally unstructured polypeptides with high binding potentials (Mollica et al.2016). Similar to other IDR-containing proteins, ORF57 NTD represents a multivalent domain allowing ORF57 to interact with target RNA transcripts via protein-RNA interaction, while simultaneously binding to other proteins that act as cofactors (Fig. 4). Formation of these ribonucleoprotein (RNP) complexes is essential for ORF57 posttranscriptional activities. Disruption of the binding sites in the ORF57 NTD abrogates ORF57 activities. The composition of ORF57-containing RNPs varies along with ongoing RNA process status and compartmentation of its RNA targets. ORF57 modulates both intron-containing and intronless RNA maturation and function by interaction with different cofactor (Boyne et al.2008; Jackson et al.2011; Majerciak et al.2008, 2011, 2014; Malik et al.2004; Massimelli et al.2011) (Fig. 5). The remaining residues (aa 220–455) of ORF57 form a structurally well-defined helix-rich (11 α-helixes) globular domain referred as the C-terminal domain (CTD) (Fig. 4). In general, two ORF57 CTDs are aligned antiparallelly as a homodimer by numerous intermolecular interactions between the globular domains of two monomers and further tightly held each other by an arm (aa 184–219) from other monomer’s NTD (Majerciak et al.2015; Yuan et al.2018). In addition, the binding of a zinc cation into the zinc-binding pocket in each monomer further stabilizes ORF57 homodimer (Fig. 4) (Yuan et al. 2018). Similar dimer structures were recently found in ORF57 homologues (Patel et al.2015; Tunnicliffe et al.2015, 2018). Functionally, the CTD dimerization is imperative for ORF57 protein stability at least partially by protecting of ORF57 from degradation by host proteasome (Majerciak and Zheng 2015).

Structural composition of ORF57 protein. KSHV ORF57 contains two structurally and functionally distinct domains. Majority of the N-terminal domain (NTD, in blue) consists of a flexible, intrinsically disordered region (IDR) that binds both target RNA (black line) and cellular cofactors (yellow circle) to form specific ribonucleoprotein complexes. The highly structured C-terminal domain (CTD, in red) contains an alpha–helix-rich globular domain and forms a homodimer stabilized by intermolecular interactions between two monomers (red and green) and Zinc cations (Zn2+) binding and then by holding each other with an arm from the NTD of other monomer (Yuan et al.2018).

A pleiotropic role of ORF57 in KSHV posttranscriptional regulation. In the nucleus ORF57 prevents hyper-polyadenylation of ORF59 through interaction with RBM15, increases stability of nuclear non-coding PAN RNA via interaction with PABPC1 and stimulates splicing of K8 pre-mRNA by interacting with SRSF3. In the cytoplasm ORF57 promotes vIL-6 translation by preventing miRNA-containing RISC complex binding to vIL-6 mRNA and blocks the formation of stress granules by binding to PKR to prevent PKR activation and phosphorylation (gray circle with P) of translational activator eIF2α.
Roles of ORF57 in Posttranscriptional Regulation
Although ORF57 is neither a transcription factor (Massimelli et al.2011), nor a bona fide RNA export factor (Pilkington et al.2012), its posttranscriptional activities are pleiotropic in nature (Majerciak and Zheng 2015) (Fig. 5). ORF57 promotes the expression of KSHV intronless mRNAs and therefore is historically referred as MTA. ORF57 interacts with cellular export factor Aly/REF and had been thought to act as a viral export factor for increased intronless RNA accumulation (Boyne et al.2008; Jackson et al.2011; Malik et al.2004). Subsequent studies showed that ORF57 interaction with Aly/REF is not a prerequisite for intronless RNA accumulation (Majerciak et al.2006a; Nekorchuk et al.2007). Instead, ORF57 accumulates the viral RNA transcripts by increasing RNA stability primarily through its two major functions: (1) ORF57 binds to the MTA-responsive element (MRE) in its target RNA, such as viral lncRNA PAN (polyadenylated nuclear RNA) and ORF59 (viral DNA polymerase processivity factor) RNA (Fig. 5), to prevent RNA decay (Majerciak et al.2006a; Massimelli et al.2011; 2015); (2) ORF57 interacts with cellular RNA-binding proteins RBM15 and OTT3 to prevent RBM15-mediated hyperpolyadenylation of ORF59 (Majerciak et al.2011) (Fig. 6). In addition, ORF57 also stabilizes the RNA transcripts of KSHV ORF47 (glycoprotein M) and ORF56 (viral primase) (Majerciak et al.2006b; Massimelli et al.2013; Pilkington et al.2012).

ORF57 prevents RBM15-mediated hyperpolyadenylation of KSHV ORF59 RNA. A A workflow of the strategy in determination of RNA poly (A) (pA) tail length (blue). First, a chimeric RNA–DNA adaptor (yellow, rUrUrUAACCGCGAATTCCAG/3AmM/-3′) was ligated to the mRNA 3′ end and followed by RT with an adaptor-specific antisene primer (5′-GACTAGCTGGAATTCGCGGTTAAA-3′). The cDNA was amplified by PCR using an ORF59-specific primer (black arrow, 5′-GGATCGTGGGAAGGTGCC-3′) in combination with an adaptor-specific primer (yellow arrow, 5′-GACTAGCTGGAATTCGCGGTTAAA-3′). The ORF59-specific primer is positioned approximately 190 bps upstream of the pA start (black double lines). The obtained PCR products (black double lines) are analyzed by Southern blot using an ORF59-specific probe (red, 5′-AATCAGGGGGTTAAATGTGGT-3′). B Length of the ORF59 RNA pA tail in HEK293T cells transfected with a vector expressing ORF59-FLAG fusion protein (pVM18) in the absence (control) or presence of ORF57 or RBM15 was determined by Southern blot as described in (A). Total RNA isolated from HEK293T cells was used in this study. The RT-PCR products of ORF59 RNA without a pA tail was about 190 bps in size and the varied sizes or smear signals of the ORF59 cDNA indicate heterogenic length of the pA tails.
KSHV ORF57 functions as a viral splicing factor and promotes RNA splicing of viral pre-mRNA transcripts in viral lytic infection (Majerciak et al.2008) (Fig. 5). As described above, almost one-third of ~90 viral genes are split genes with one or more introns, of which RNA splicing is a vital part of their posttranscriptional processing (Arias et al.2014; Sharp et al.2002; Sturzl et al.1999; Tang and Zheng 2002; Yamanegi et al.2005; Zheng 2003). In this regard, RTA, K8 and K8.1 are transcribed, respectively, as a tricistronic RTA RNA with 4 introns, bicistronic K8 RNA with 3 introns and monocistronic K8.1 RNA with one intron (Tang and Zheng 2002). All of these three gene transcripts are polyadenylated by using a common pA site downstream of the K8.1 ORF and thus, both K8 and K8.1 coding regions are the 3′ UTR region of RTA. ORF57 binds the K8 intron 2 (RTA intron 3), host splicing factors and nuclear small RNAs to promote splicing of RTA and K8 pre-mRNA transcripts (Majerciak et al.2008, 2014). This stimulatory effect of ORF57 on splicing of this intron depends on its interaction with the cellular splicing factor SRSF3 (or SRp20) which binds to this intron and blocks it from splicing (Majerciak et al.2014) (Fig. 5).
KSHV ORF57 interferes with host miRNA machinery and regulates RNA stability and translation (Fig. 5). This function of ORF57 is two-fold: binding to miRNA seed matches (binding sites) in the target RNAs and interacting with Ago2 and GW182, two major components of RISC (RNA-induced silencing complex). ORF57 binds to a miR-1293 binding site in the ORF region of viral IL-6 RNA and a miR-608 binding site in the ORF region of human IL-6 RNA to prevent miRNA-mediated inhibition on IL-6 translation (Kang JG et al. 2011a, b). ORF57 also binds Ago2 and GW182 to prevent Ago2-GW182 interaction and inhibits the formation of RNA processing bodies (P-bodies) (Sharma N et al. manuscript in submission). Another function of ORF57 in promotion of viral protein translation is to suppress stress granule formation during lytic infection by interacting with a double-strand RNA-binding protein kinase R (PKR) and blocking PKR activation and phosphorylation of eIF2α, an eukaryotic translation initiation factor (Sharma et al.2017).
KSHV Life Cycle and Oncogenesis
The close association of KSHV with some human malignancies (KS, PEL and MCD) has caused virologists to consider KSHV as a human oncovirus. The highlights of KSHV infection are a higher cell growth rate with an extended lifespan, altered cell morphology, deregulated angiogenesis, elevated inflammation and immune evasion to support tumor growth (Dittmer and Damania 2016; Fatahzadeh 2012; Kaplan 2013; Radu and Pantanowitz 2013). The lack of an in vitro KSHV cellular transformation model and KS cell lines has hindered research on the mechanisms of KSHV-induced cellular transformation and oncogenesis. Recent progress has been made in KSHV immortalization and transformation of primary rat embryonic metanephric mesenchymal precursor (MM) cells and demonstrated that KSHV-transformed MM cells (KMM) efficiently induce tumors with pathological features similar to those of KS patients (Jones et al.2014; Moody et al.2013). Humanized BLT (bone marrow, liver, and thymus) mice infected by inoculation with KSHV.219 virus via the oral and vaginal routes could be a useful model for understanding the pathogenesis and transmission of KSHV (Wang et al.2014). Further, more evidences have been acquired that KSHV can also infect and induce pathological phenotypes in human mesenchymal stem cells, which make the cells as potential origin of KS tumor cells apart from the previous accepted endothelial cell lineage (Lee et al.2016c; Li et al.2018).
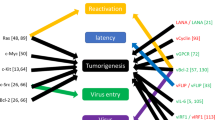
The evidence that most KS spindle cells are latently infected with KSHV suggests that latency may contribute to KSHV-induced malignancies and pathogenesis. Moreover, a small group of cells with latent KSHV infection has been found undergoing lytic replication and producing virus, which suggests that lytic replication may also be important for tumor development (Fig. 7). Thus, most likely, both latent and lytic phases of the virus life cycle are involved in tumor initiation and progression, but their underlying mechanisms could be very different.

A brief summary of the possible mechanisms of KSHV-mediated pathogenesis at the cellular level. Shown in the diagram are viral lytic proteins in orange and viral latent proteins in green. (i) KSHV vGPCR, K1, K15, and vIL-6 proteins activate the PI3 Kγ/AKT, MAPK, JAK/STAT, NF-κB, MEK/ERK/JNK signaling pathways for infected cells to secrete a number of cytokines and chemokines, such as IL-6/8/10/1β, GM-CSF and VEGF; KSHV miR-K5/9/10a also induced the cytokines secretion. miR-K2/3/5/6 can induce angiogenesis-related factors, like VEGFA; (ii) KSHV encodes three chemokine homologs: viral CC-chemokine ligand-1-3 (vCCL1-K6, vCCL2-K4 and vCCL3-K4.1) to downregulate cellular immune response; (iii) KSHV RTA up-regulates the Notch ligand JAG1 by interacting with LEF1 and triggers Notch activation in neighboring cells. The activated Notch inhibits KSHV reactivation in those neighboring cells. It provides an insight into the mechanism by which a minority of viruses undergoes reactivation, while the majority maintains a persistent latent infection in KS tissues; (iv) v-FLIP in the infected cells inhibits the activation of pro-caspase-8 and has the ability to induce the expression of anti-apoptotic proteins via activation of NF-κB to upregulate pro-survival genes expression. vBcl-2, a homolog of cellular Bcl-2, inhibits apoptosis via tightly binding proapoptotic peptides. KSHV miR-K10a blocks TWEAK-induced apoptosis; (v) LANA affects the BMP signaling pathway and converts it to an oncogenic BMP-Smad1-Id pathway, which might contribute to the pathogenesis of KSHV-induced malignancies; (vi) KSHV LANA binds to and blocks p53-transcriptional activity to inhibit p53-induced cell death. LANA also binds and inactivates pRb, thereby removing the inhibition of pRb-induced cell cycle arrest. LANA binds to GSK-3β and thus accumulates unphosphorylated β-catenin, which in turn affects the cell cycle. v-Cyclin regulates the cell cycle and cell proliferation by constitutive activation of cellular cyclin-dependent kinase 6. vIRF-3 acts as a transcriptional activator on genes controlled by cellular IRF-3 and IRF-7. Further, vIRF-3 negatively regulates p53 protein stability, thereby inhibits p53-mediated activation of p21 gene transcription.
KSHV infection can elicit many cellular pathways to facilitate cell survival and proliferation and thus promotes tumor development. LANA, the most important latent protein that is essential for virus maintenance, has been shown to interact with p53 and Rb, the common tumor suppressors (Rivas et al.2001; Shin et al.2006). This interaction inhibits cell death and the cell cycle checkpoint, thus contributing to KSHV-induced oncogenesis. KSHV v-Cyclin, another latent protein, has been demonstrated to accelerate cell proliferation by binding to phosphorylated CDK6 (Sarek et al.2006).
To better maintain tumor growth, KSHV has also evolved strategies to avoid the apoptosis of dysregulated cellular pathways. Many studies have found that v-FLIP, a latent protein, targets the classical NF-κB pathway to promote tumor cell survival. Moreover, NF-κB activation not only leads to cellular transformation, but also increases the incidence of lymphoma in transgenic v-FLIP mice (Guasparri et al.2004). The inhibition of the NF-κB pathway using specific inhibitors was found to suppress tumor growth in mouse models and in tissue culture in vitro (Grossmann et al.2006; Matta et al.2003; Ye et al.2008).
KS is a highly angiogenic neoplasm characterized by heavily and irregularly shaped blood vessels, indicating that during KSHV infection, many angiogenic and lymphangiogenic factors are produced at a relatively high level (Boshoff 2002; Orenstein 2008). Unlike the general action of the host, pathological angiogenesis is correlated with tumor growth and metastasis (O’Byrne et al.2000; Sakakibara and Tosato 2009), and while the mechanisms of angiogenesis in KS tumor development are not yet clearly understood, KSHV-induced angiogenic factors and inflammatory cytokines appear to be essential for driving KS tumor development (Dimaio and Lagunoff 2012; Purushothaman et al.2016). The angiogenic cytokines IL-6, IL-8, basic fibroblast growth factor (b-FGF), ephrin B2, cyclooxygenase-2 (Cox-2), heme oxygenase-1(HO-1), Ang2 and MMPs could be induced after KSHV infection (Botto et al.2015; Cianfrocca et al.2002; Dai et al.2016; Haddad et al.2008; McAllister et al.2004; Meade-Tollin et al.1999; Paul et al.2013; Sadagopan et al.2009; Sharma-Walia et al.2010; Ye et al.2013). Additionally, a number of KSHV-encoded proteins, such as vIL-6, vCCL-1, vCCL-2 and vGPCR, have been shown to act together with vIRF1, v-FLIP, v-Cyclin to stimulate angiogenesis (Asou et al.1998; Jones et al.1999; Wu et al.2014). Furthermore, KSHV-encoded miRNAs could induce angiogenesis by activating multiple cellular signaling (Li et al.2016c, 2017; Ramalingam et al.2015).
To avoid attack from cellular factors, KSHV exploits immune evasion strategies to promote tumorigenesis (Moore and Chang 2003). KSHV encodes multiple proteins which directly or indirectly inhibit cellular immune-related processes and signaling, such as cytokine secretion, antigen processing and presentation, interferon signaling and complement system (Cai et al.2010). KSHV encodes viral IL-6, which directly binds to gp130, and activates MAPK serine/threonine kinase pathways (Cousins and Nicholas 2013; Molden et al.1997). To interfere with the complement system, ORF4, also called complement control protein (KCP), blocks progression of the complement cascade (Okroj et al.2011; Spiller et al.2003). KSHV encodes four viral homologs of IRF (vIRF1–4) (Baresova et al.2013). vIRF1 represses cellular IFN-mediated signal transduction by directly interacting with the IFN-stimulated response DNA element and contributes to cell transformation in nude mice to promote tumorigenesis (Baresova et al.2013). Another important immune evasion strategy exploited by KSHV is downregulation of cell surface MHC class I molecules, and ORF K3 (MIR1) and ORF K5 (MIR2) modulate MHC I molecules on infected cell surfaces via ubiquitylation by acting as E3 ligases (Brulois et al.2014; Ishido et al.2000). KSHV encodes several viral chemokines (vCCL) to evade adaptive immunity (Hu and Usherwood 2014). KSHV modulates Toll-like receptor signaling and PKR pathways to disrupt the innate immunity response to virus infection (Lee et al.2016b; Sharma et al.2017). Recently, several reports showed that KSHV encodes ORF52, an abundant tegument protein, which directly binds to cytosolic DNA sensor cGAS subverting its enzymatic activity, thus enabling KSHV to escape the host immune response (Li et al.2016a; Wu et al.2015) (Fig. 7).
Perspectives
More than two decades after KSHV discovery, the joint efforts of scientists worldwide have propelled numerous advances in every aspect of KSHV research, but many questions obviously remain. Up to date, multiple KSHV genomes from infected B cells, tumor tissues, purified virions and BAC-derived genomes have been sequenced and ~ 81 ORFs have been roughly annotated to a ~137-kb long unique region (LUR) according to a continuous stretch of > 100 codons that begins with a start codon (usually AUG) and ends at a stop codon (usually UAA, UAG or UGA). Although this ORF annotation was initially useful for our quick start to understand the functions of individual viral proteins by using an overexpression system, the caveat is that KSHV bears a complex genome having many split genes and the current ORF annotation completely ignored the discontinuous feature of these split genes in the KSHV genome. Thus, the published studies based on the initial annotated ORFs on split genes were not reliable and misleading the field. Moreover, many annotated genes are clustered in a locus to share a common pA site downstream for their expression (Majerciak et al.2013) and the annotated ORF(s) downstream in the gene cluster locus could be the 3′ untranslated region (UTR) of other gene (s) upstream. A special precaution has to pay on these gene structures and expression, especially when siRNA or gene knockout approaches are chosen. Repeatedly, we saw many laboratories publishing their studies by using siRNA or gene knockout technologies to target their favorable gene which is also the 3′ UTR region (s) of other gene (s) to draw their “big” conclusion of the targeted gene functions.
Development of the first B cell lines from PEL patients allowed analysis of the viral genome and different aspects of the viral life cycle (Renne et al.1996b). Using purified and concentrated KSHV particles, primary endothelial cells had been found to be infected and transformed at the presence of vascular endothelial growth factor A (VEGF) (Flore et al.1998). Limited by the PEL cells origin, the impact of viral replication on endothelial function or proliferation hadn’t been well studied. Telomerase-immortalized microvascular endothelial cells (TIME) with KSHV virions had been found to support efficient latent infection and reagents-induced lytic growth and infectious progeny generation (Lagunoff et al.2002). To further elucidate details of the virus life cycle, especially the lytic phase, under relatively physiological conditions, a tetracycline-inducible expression system (TREx BCBL1-Rta) was developed in KSHV-infected PEL cells (Nakamura et al.2003). Another cell line, iSLK.219 cells, was developed to study latent viral gene expression with tightly controlled and inducible reactivation machinery (Myoung and Ganem 2011a). Moreover, manipulation of the viral genome using the BAC-mediated recombinant system has become more efficient and maneuverable, thus making in-depth functional studies on viral genes and virus-host interactions possible (Brulois et al.2012; Zhou et al.2002). Results obtained using KSHV-transformed MM cells suggest that these cells are useful for studying mechanisms of KSHV-induced growth deregulation and oncogenesis (Jones et al.2012; Moody et al.2013). To study the viral pathogenesis in vivo, many methods have been utilized to develop animal models ranging from severe combined immunodeficient (SCID) mice to Rhesus macaques, marmosets, and mice (Chang et al.2009; Picchio et al.1997; Renne et al.2004). However, due to the host specificity of the virus, attempts to develop a better system are continuing.
Although these powerful tools have been developed and the regulatory mechanisms underlying the KSHV life cycle and related pathogenesis have become increasingly clear, many questions need to be addressed. For example, why is latency the default viral gene expression pattern after KSHV primary infection? What detailed and precise regulatory events occurring during the early stage of infection contribute to establishing the so-called default latency? What is the basis for a relatively active viral gene expression pattern instead of typical latency in certain cell types? What are the intrinsic physiological stimuli triggering lytic reactivation from latency? We believe that answering these questions will help better understand regulation of viral life cycles and related pathogenesis, and eventually benefit the development of new therapeutic strategies for KSHV-associated diseases.
References
Abend JR, Uldrick T, Ziegelbauer JM (2010) Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi’s sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J Virol 84:12139–12151
Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM (2012) Kaposi’s sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88, two components of the toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J Virol 86:11663–11674
Adams V, Kempf W, Hassam S, Briner J, Schmid M, Moos R, Pfaltz M (1995) Detection of several types of human papilloma viruses in AIDS-associated Kaposi’s sarcoma. J Med Virol 46:189–193
Ajiro M, Zheng ZM (2014) Oncogenes and RNA splicing of human tumor viruses. Emerg Microbes Infect 3:e63
Akula SM, Pramod NP, Wang FZ, Chandran B (2001) Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 284:235–249
Akula SM, Pramod NP, Wang F, Chandran B (2002) Integrin α3/β1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407–419
Al-Kzayer LF, Keizer P, Abdulraheem FT, Sano K, Kamata M, Sakashita K, Habbaba LA, Koike K (2016) Rapidly progressive Kaposi’s Sarcoma in an Iraqi boy received valproic acid: a case report and review of literature. BMC Pediatr 16:111
Ameres SL, Zamore PD (2013) Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14:475–488
Andrei G, Snoeck R (2015) Kaposi’s sarcoma-associated herpesvirus: the role of lytic replication in targeted therapy. Curr Opin Infect Dis 28:611–624
Areste C, Blackbourn DJ (2009) Modulation of the immune system by Kaposi’s sarcoma-associated herpesvirus. Trends Microbiol 17:119–129
Arias C, Welsberd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, Holdorf M, Weissman JS, Ganem D (2014) KSHV 2.0: a comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog 10:e1003847
Asou H, Said JW, Yang R, Munker R, Park DJ, Kamada N, Koeffler HP (1998) Mechanisms of growth control of Kaposi’s sarcoma–associated herpes virus–associated primary effusion lymphoma cells. Blood 91:2475–2481
Avitabile E, Forghieri C, Campadelli-Fiume G (2009) Cross talk among the glycoproteins involved in herpes simplex virus entry and fusion: the interaction between gB and gH/gL does not necessarily require gD. J Virol 83:10752–10760
Baghian A, Luftig M, Black JB, Meng YX, Pau CP, Voss T, Pellett PE, Kousoulas KG (2000) Glycoprotein B of human herpesvirus 8 is a component of the virion in a cleaved form composed of amino- and carboxyl-terminal fragments. Virology 269:18–25
Bagneris C, Ageichik AV, Cronin N, Wallace B, Collins M, Boshoff C, Waksman G, Barrett T (2008) Crystal structure of a vFlip-IKKgamma complex: insights into viral activation of the IKK signalosome. Mol Cell 30:620–631
Bajaj BG, Verma SC, Lan K, Cotter MA, Woodman ZL, Robertson ES (2006) KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology 351:18–28
Ballestas ME, Kaye KM (2001) Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J Virol 75:3250–3258
Ballestas ME, Chatis PA, Kaye KM (1999) Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644
Baresova P, Pitha PM, Lubyova B (2013) Distinct roles of Kaposi’s sarcoma-associated herpesvirus-encoded viral interferon regulatory factors in inflammatory response and cancer. J Virol 87:9398–9410
Bechtel JT, Winant RC, Ganem D (2005) Host and viral proteins in the virion of Kaposi’s sarcoma-associated herpesvirus. J Virol 79:4952–4964
Bellare P, Ganem D (2009) Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6:570–575
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326
Bieleski L, Talbot SJ (2001) Kaposi’s sarcoma-associated herpesvirus vCyclin open reading frame contains an internal ribosome entry site. J Virol 75:1864–1869
Boldogh I, Szaniszlo P, Bresnahan WA, Flaitz CM, Nichols MC, Albrecht T (1996) Kaposi’s sarcoma herpesvirus-like DNA sequences in the saliva of individuals infected with human immunodeficiency virus. Clin Infect Dis 23:406–407
Boshoff C (2002) Kaposi’s Sarcoma Biology. IUBMB Life 53:259–261
Botto S, Totonchy JE, Gustin JK, Moses AV (2015) Kaposi sarcoma herpesvirus induces HO-1 during de novo infection of endothelial cells via viral miRNA-dependent and -independent mechanisms. MBio 6:e00668
Boyne JR, Colgan KJ, Whitehouse A (2008) Recruitment of the complete hTREX complex is required for Kaposi’s sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog 4:e1000194
Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU (2012) Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720
Brulois K, Toth Z, Wong LY, Feng P, Gao SJ, Ensser A, Jung JU (2014) Kaposi’s sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J Virol 88:9335–9349
Bu W, Palmeri D, Krishnan R, Marin R, Aris VM, Soteropoulos P, Lukac DM (2008) Identification of direct transcriptional targets of the Kaposi’s sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J Virol 82:10709–10723
Byun H, Gwack Y, Hwang S, Choe J (2002) Kaposi’s Sarcoma-associated herpesvirus open reading frame (ORF) 50 transactivates K8 and ORF57 promoters via heterogeneous response elements. Mol Cells 14:185–191
Cai X, Cullen BR (2006) Transcriptional origin of Kaposi’s sarcoma-associated herpesvirus microRNAs. J Virol 80:2234–2242
Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR (2005) Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci USA 102:5570–5575
Cai Q, Knight JS, Verma SC, Zald P, Robertson ES (2006) EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog 2:1002–1012
Cai Q, Verma SC, Lu J, Robertson ES (2010) Molecular biology of Kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv Virus Res 78:87–142
Campbell M, Izumiya Y (2012) Post-translational modifications of Kaposi’s sarcoma-associated herpesvirus regulatory proteins-SUMO and KSHV. Front Microbiol 3:31
Campbell M, Chang PC, Huerta S, Izumiya C, Davis R, Tepper CG, Kim KY, Shevchenko B, Wang DH, Jung JU, Luciw PA, Kung HJ, Izumiya Y (2012) Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J Biol Chem 287:5806–5818
Cao Y, Minhas V, Tan X, Huang J, Wang B, Zhu M, Gao Y, Zhao T, Yang L, Wood C (2014) High prevalence of early childhood infection by Kaposi’s sarcoma-associated herpesvirus in a minority population in China. Clin Microbiol Infect 20:475–481
Carroll KD, Khadim F, Spadavecchia S, Palmeri D, Lukac DM (2007) Direct interactions of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J Virol 81:8451–8467
Chandriani S, Ganem D (2010) Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J Virol 84:5565–5573
Chang PC, Li M (2008) Kaposi’s sarcoma-associated herpesvirus K-cyclin interacts with Cdk9 and stimulates Cdk9-mediated phosphorylation of p53 tumor suppressor. J Virol 82:278–290
Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–1869
Chang H, Wachtman LM, Pearson CB, Lee JS, Lee HR, Lee SH, Vieira J, Mansfield KG, Jung JU (2009) Non-human primate model of Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog 5:e1000606
Chang HC, Hsieh TH, Lee YW, Tsai CF, Tsai YN, Cheng CC, Wang HW (2016) c-Myc and viral cofactor Kaposin B co-operate to elicit angiogenesis through modulating miRNome traits of endothelial cells. BMC Syst Biol 10(Suppl 1):1
Chaudhary PM, Jasmin A, Eby MT, Hood L (1999) Modulation of the NF-κB pathway by virally encoded death effector domains-containing proteins. Oncogene 18:5738–5746
Chen J, Ueda K, Sakakibara S, Okuno T, Parravicini C, Corbellino M, Yamanishi K (2001) Activation of latent Kaposi’s sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc Natl Acad Sci USA 98:4119–4124
Chen J, Ye F, Xie J, Kuhne K, Gao SJ (2009) Genome-wide identification of binding sites for Kaposi’s sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 386:290–302
Chen HS, Wikramasinghe P, Showe L, Lieberman PM (2012) Cohesins repress Kaposi’s sarcoma-associated herpesvirus immediate early gene transcription during latency. J Virol 86:9454–9464
Chen HS, Lu F, Lieberman PM (2013) Epigenetic regulation of EBV and KSHV latency. Curr Opin Virol 3:251–259
Cheng F, Weidner-Glunde M, Varjosalo M, Rainio EM, Lehtonen A, Schulz TF, Koskinen PJ, Taipale J, Ojala PM (2009) KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog 5:e1000324
Chugh PE, Sin SH, Ozgur S, Henry DH, Menezes P, Griffith J, Eron JJ, Damania B, Dittmer DP (2013) Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog 9:e1003484
Cianfrocca M, Cooley TP, Lee JY, Rudek MA, Scadden DT, Ratner L, Pluda JM, Figg WD, Krown SE, Dezube BJ (2002) Matrix metalloproteinase inhibitor COL-3 in the treatment of AIDS-related Kaposi’s sarcoma: a phase I AIDS malignancy consortium study. J Clin Oncol 20:153–159
Cook-Mozaffari P, Newton R, Beral V, Burkitt DP (1998) The geographical distribution of Kaposi’s sarcoma and of lymphomas in Africa before the AIDS epidemic. Br J Cancer 78:1521–1528
Corcoran JA, McCormick C (2015) Viral activation of stress-regulated Rho-GTPase signaling pathway disrupts sites of mRNA degradation to influence cellular gene expression. Small GTPases 6:178–185
Coscoy L (2007) Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Nat Rev Immunol 7:391–401
Cousins E, Nicholas J (2013) Role of human herpesvirus 8 interleukin-6-activated gp130 signal transducer in primary effusion lymphoma cell growth and viability. J Virol 87:10816–10827
Dai L, Qiao J, Nguyen D, Struckhoff AP, Doyle L, Bonstaff K, Valle LD, Parsons C, Toole BP, Renne R, Qin Z (2016) Role of heme oxygenase-1 in the pathogenesis and tumorigenicity of Kaposi’s sarcoma-associated herpesvirus. Oncotarget 7:10459–10471
Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R (2001) Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 97:3244–3250
Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jager S, Shales M, Horner J, Hernandez RD, Krogan NJ, Glaunsinger BA (2015) Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57:349–360
De Leon Vazquez E, Kaye KM (2011) The internal Kaposi’s sarcoma-associated herpesvirus LANA regions exert a critical role on episome persistence. J Virol 85:7622–7633
Deng H, Young A, Sun R (2000) Auto-activation of the rta gene of human herpesvirus-8/Kaposi’s sarcoma-associated herpesvirus. J Gen Virol 81:3043–3048
Deng Z, Wang Z, Lieberman PM (2012) Telomeres and viruses: common themes of genome maintenance. Front Oncol 2:201
Dimaio TA, Lagunoff M (2012) KSHV induction of angiogenic and lymphangiogenic phenotypes. Front Microbiol 3:102
Dittmer DP, Damania B (2016) Kaposi sarcoma-associated herpesvirus: immunobiology, oncogenesis, and therapy. J Clin Invest 126:3165–3175
Dittmer D, Lagunoff M, Renne R, Staskus KA, Haase A, Ganem D (1998) A cluster of latently expressed genes in Kaposi’s Sarcoma-associated herpesvirus. J Virol 72:8309–8315
Dollard SC, Nelson KE, Ness PM, Stambolis V, Kuehnert MJ, Pellett PE, Cannon MJ (2005) Possible transmission of human herpesvirus-8 by blood transfusion in a historical United States cohort. Transfusion 45:500–503
Dollard SC, Butler LM, Jones AM, Mermin JH, Chidzonga M, Chipato T, Shiboski CH, Brander C, Mosam A, Kiepiela P, Hladik W, Martin JN (2010) Substantial regional differences in human herpesvirus 8 seroprevalence in sub-Saharan Africa: insights on the origin of the “Kaposi’s sarcoma belt”. Int J Cancer 127:2395–2401
Dorsett D (2011) Cohesin: genomic insights into controlling gene transcription and development. Curr Opin Genet Dev 21:199–206
Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM (2003) Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) epidemiology and pathogenesis. Microbiol Mol Biol Rev 67:175–212
Duan W, Wang S, Liu S, Wood C (2001) Characterization of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8 ORF57 promoter. Arch Virol 146:403–413
Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, Marck EV, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C (1999) Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc Natl Acad Sci USA 96:4546–4551
Ellison TJ, Izumiya Y, Izumiya C, Luciw PA, Kung HJ (2009) A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi’s sarcoma-associated herpesvirus. Virology 387:76–88
Eltom MA, Mbulaiteye SM, Dada AJ, Whitby D, Biggar RJ (2002) Transmission of human herpesvirus 8 by sexual activity among adults in Lagos, Nigeria. AIDS 16:2473–2478
Fakhari FD, Dittmer DP (2002) Charting latency transcripts in Kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol 76:6213–6223
Fatahzadeh M (2012) Kaposi sarcoma: review and medical management update. Oral Surg Oral Med Oral Pathol Oral Radiol 113:2–16
Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M (2003) KSHV vFLIP binds to IKK-γ to activate IKK. J Cell Sci 116:3721–3728
Flore O, Raffi S, Ely S, O’Leary JJ, Hyjek EM, Cesarman E (1998) Transformation of primary human endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Nature 394:588–592
Ford PW, Bryan BA, Dyson OF, Weidner DA, Chintalgattu V, Akula SM (2006) Raf/MEK/ERK signalling triggers reactivation of Kaposi’s sarcoma-associated herpesvirus latency. J Gen Virol 87:1139–1144
Forte E, Raja AN, Shamulailatpam P, Manzano M, Schipma MJ, Casey JL, Gottwein E (2015) MicroRNA-mediated transformation by the Kaposi’s sarcoma-associated herpesvirus Kaposin locus. J Virol 89:2333–2341
Fu B, Sun F, Li B, Yang L, Zeng Y, Sun X, Xu F, Rayner S, Guadalupe M, Gao SJ, Wang L (2009) Seroprevalence of Kaposi’s sarcoma-associated herpesvirus and risk factors in Xinjiang, China. J Med Virol 81:1422–1431
Fujimuro M, Liu J, Zhu J, Yokosawa H, Hayward SD (2005) Regulation of the interaction between glycogen synthase kinase 3 and the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen. J Virol 79:10429–10441
Full F, Jungick D, Reuter N, Bogner E, Brulois K, Scholz B, Sturzl M, Myoung J, Jung JU, Stamminger T, Ensser A (2014) Kaposi’s sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-Instituted intrinsic immunity. PLoS Pathog 10:e1003863
Ganem D (1997) KSHV and Kaposi’s Sarcoma: the end of the beginning? Cell 91:157–160
Ganem D (2010) KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120:939–949
Gao SJ, Kingsley L, Li M, Zheng W, Parravicini C, Ziegler J, Newton R, Rinaldo CR, Saah A, Phair J, Detels R, Chang Y, Moore PS (1996) KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat Med 2:925–928
Garber AC, Shu MA, Hu J, Renne R (2001) DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J Virol 75:7882–7892
Giffin L, Damania B (2014) KSHV: pathways to tumorigenesis and persistent infection. Adv Virus Res 88:111–159
Gillen J, Li W, Liang Q, Avey D, Wu J, Wu F, Myoung J, Zhu F (2015) A survey of the interactome of Kaposi’s sarcoma-associated herpesvirus ORF45 revealed its binding to viral ORF33 and cellular USP7, resulting in stabilization of ORF33 that is required for production of progeny viruses. J Virol 89:4918–4931
Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA, Mittnacht S (1997) The cyclin encoded by Kaposi’s sarcoma-associated herpesvirus stimulates cdk6 To phosphorylate the retinoblastoma protein and histone H1. J Virol 71:4193–4198
Gottwein E, Cai X, Cullen BR (2006) Expression and function of microRNAs encoded by Kaposi’s sarcoma-associated herpesvirus. Cold Spring Harb Symp Quant Biol 71:357–364
Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutschek J, Ohler U, Cullen BR (2007) A viral microRNA functions as an orthologue of cellular miR-155. Nature 450:1096–1099
Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, Shamulailatpam P, Love CL, Dave SS, Tuschl T, Ohler U, Cullen BR (2011) Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10:515–526
Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G (2000) Kaposi’s sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J Virol 74:6207–6212
Graham C, Matta H, Yang Y, Yi H, Suo Y, Tolani B, Chaudhary PM (2013) Kaposi’s sarcoma-associated herpesvirus oncoprotein K13 protects against B cell receptor-induced growth arrest and apoptosis through NF-kappaB activation. J Virol 87:2242–2252
Gregory SM, West JA, Dillon PJ, Hilscher C, Dittmer DP, Damania B (2009) Toll-like receptor signaling controls reactivation of KSHV from latency. Proc Natl Acad Sci USA 106:11725–11730
Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, Ting JP, Damania B (2011) Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331:330–334
Griffiths R, Whitehouse A (2007) Herpesvirus saimiri episomal persistence is maintained via interaction between open reading frame 73 and the cellular chromosome-associated protein MeCP2. J Virol 81:4021–4032
Grossmann C, Podgrabinska S, Skobe M, Ganem D (2006) Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J Virol 80:7179–7185
Grundhoff A, Sullivan CS, Ganem D (2006) A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 12:733–750
Guasparri I, Keller SA, Cesarman E (2004) KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med 199:993–1003
Gunther T, Grundhoff A (2010) The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog 6:e1000935
Haas DA, Bala K, Busche G, Weidner-Glunde M, Santag S, Kati S, Gramolelli S, Damas M, Dittrich-Breiholz O, Kracht M, Ruckert J, Varga Z, Keri G, Schulz TF (2013) The inflammatory kinase MAP4K4 promotes reactivation of Kaposi’s sarcoma herpesvirus and enhances the invasiveness of infected endothelial cells. PLoS Pathog 9:e1003737
Haddad L, El Hajj H, Abou-Merhi R, Kfoury Y, Mahieux R, El-Sabban M, Bazarbachi A (2008) KSHV-transformed primary effusion lymphoma cells induce a VEGF-dependent angiogenesis and establish functional gap junctions with endothelial cells. Leukemia 22:826–834
Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, Holzer A, Schmidt M, Chen J, Konig S, Ensser A, Myoung J, Brockmeyer NH, Sturzl M, Fleckenstein B, Neipel F (2012) The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat Med 18:961–966
Han Z, Swaminathan S (2006) Kaposi’s sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J Virol 80:5251–5260
Haque M, Davis DA, Wang V, Widmer I, Yarchoan R (2003) Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) contains hypoxia response elements: relevance to lytic Induction by hypoxia. J Virol 77:6761–6768
Hensler HR, Tomaszewski MJ, Rappocciolo G, Rinaldo CR, Jenkins FJ (2014) Human herpesvirus 8 glycoprotein B binds the entry receptor DC-SIGN. Virus Res 190:97–103
Herold M, Bartkuhn M, Renkawitz R (2012) CTCF: insights into insulator function during development. Development 139:1045–1057
Hladik W, Dollard SC, Mermin J, Fowlkes AL, Downing D, Amin MM, Banage F, Nzaro E, Kataaha P, Dondero TJ, Pellett PE, Lackritz EM (2006) Transmission of human herpesvirus 8 by blood transfusion. N Engl J Med 355:1331–1338
Hladik W, Pellett PE, Hancock J, Downing R, Gao H, Packel L, Mimbe D, Nzaro E, Mermin J (2012) Association between transfusion with human herpesvirus 8 antibody-positive blood and subsequent mortality. J Infect Dis 206:1497–1503
Hu J, Renne R (2005) Characterization of the minimal replicator of Kaposi’s sarcoma-associated herpesvirus latent origin. J Virol 79:2637–2642
Hu Z, Usherwood EJ (2014) Immune escape of gamma-herpesviruses from adaptive immunity. Rev Med Virol 24:365–378
Hu J, Yang Y, Turner PC, Jain V, McIntyre LM, Renne R (2014) LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog 10:e1004240
Hu M, Wang C, Li W, Lu W, Bai Z, Qin D, Yan Q, Zhu J, Krueger BJ, Renne R, Gao SJ, Lu C (2015) A KSHV microRNA directly targets G protein-coupled receptor kinase 2 to promote the migration and invasion of endothelial cells by inducing CXCR113 and activating AKT signaling. PLoS Pathog 11:e1005171
Hunte R, Alonso P, Thomas R, Bazile CA, Ramos JC, van der Weyden L, Dominguez-Bendala J, Khan WN, Shembade N (2018) CADM1 is essential for KSHV-encoded vGPCR-and vFLIP-mediated chronic NF-kappaB activation. PLoS Pathog 14:e1006968
Iscovich J, Boffetta P, Winkelmann R, Brennan P, Azizi E (1998) Classic Kaposi’s sarcoma in Jews living in Israel. AIDS 12:2067–2072
Ishido S, Wang C, Lee BS, Cohen GB, Jung JU (2000) Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J Virol 74:5300–5309
Izumiya Y, Lin SF, Ellison TJ, Levy AM, Mayeur GL, Izumiya C, Kung HJ (2003) Cell cycle regulation by Kaposi’s Sarcoma-associated herpesvirus K-bZIP: direct interaction with cyclin-CDK2 and induction of G1 growth arrest. J Virol 77:9652–9661
Jackson BR, Boyne JR, Noerenberg M, Taylor A, Hautbergue GM, Walsh MJ, Wheat R, Blackbourn DJ, Wilson SA, Whitehouse A (2011) An interaction between KSHV ORF57 and UIF provides mRNA-adaptor redundancy in herpesvirus intronless mRNA export. PLoS Pathog 7:e1002138
Jarviluoma A, Koopal S, Rasanen S, Makela TP, Ojala PM (2004) KSHV viral cyclin binds to p27KIP1 in primary effusion lymphomas. Blood 104:3349–3354
Jenner RG, Alba MM, Boshoff C, Kellam P (2001) Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J Virol 75:891–902
Jha HC, Lu J, Verma SC, Banerjee S, Mehta D, Robertson ES (2014) Kaposi’s sarcoma-associated herpesvirus genome programming during the early stages of primary infection of peripheral blood mononuclear cells. MBio 5:e02261-14
Jones KD, Aoki Y, Chang Y, Moore PS, Yarchoan R, Tosato G (1999) Involvement of Interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi’s sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood 94:2871–2879
Jones T, Ye F, Bedolla R, Huang Y, Meng J, Qian L, Pan H, Zhou F, Moody R, Wagner B, Arar M, Gao SJ (2012) Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. J Clin Invest 122:1076–1081
Jones T, Ramos da Silva S, Bedolla R, Ye F, Zhou F, Gao SJ (2014) Viral cyclin promotes KSHV-induced cellular transformation and tumorigenesis by overriding contact inhibition. Cell Cycle 13:845–858
Jr JF, Kong WP, Hottiger MO, Nabel GJ (1999) p53 inhibition by the LANA of KSHV protect against cell death. Nature 402:889–894
Kakavand-Ghalehnoei R, Shoja Z, Najafi A, Mollahoseini MH, Shahmahmoodi S, Marashi SM, Nejati A, Jalilvand S (2016) Prevalence of human herpesvirus-8 among HIV-infected patients, intravenous drug users and the general population in Iran. Sex Health 13:295–298
Kaleeba JA, Berger EA (2006) Kaposi’s sarcoma–associated Herpesvirus fusion-entry receptor: cystine transporter xCT. Science 311:1921–1924
Kang H, Wiedmer A, Yuan Y, Robertson ES, Lieberman PM (2011) Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog 7:e1002140
Kang JG, Majerciak V, Uldrick TS, Wang X, Kruhlak M, Yarchoan R, Zheng ZM (2011a) Kaposi’s sarcoma-associated herpesviral IL-6 and human IL-6 open reading frames contain miRNA binding sites and are subject to cellular miRNA regulation. J Pathol 225:378–389
Kang JG, Pripuzova N, Majerciak V, Kruhlak M, Le SY, Zheng ZM (2011b) Kaposi’s sarcoma-associated herpesvirus ORF57 promotes escape of viral and human interleukin-6 from microRNA-mediated suppression. J Virol 85:2620–2630
Kaplan L (2013) Human herpesvirus-8: Kaposi sarcoma, multicentric castleman disease, and primary effusion lymphoma. Hematol Am Soc Hematol Educ Program 2013:103–108
Kedes DH, Lagunoff M, Renne R, Ganem D (1997) Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi’s Sarcoma-associated Herpesvirus. J Clin Investig 100:2606–2610
Kelley-Clarke B, De Leon-Vazquez E, Slain K, Barbera AJ, Kaye KM (2009) Role of Kaposi’s sarcoma-associated herpesvirus C-terminal LANA chromosome binding in episome persistence. J Virol 83:4326–4337
Kieffer-Kwon P, Happel C, Uldrick TS, Ramalingam D, Ziegelbauer JM (2015) KSHV microRNAs repress tropomyosin 1 and increase anchorage-independent growth and endothelial tube formation. PLoS ONE 10:e0135560
Kieshner JR, Lukac DM, Chang J, Ganem D (2000) Kaposi’s sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J Virol 74:3586–3597
Kliche S, Nagel W, Kremmer E, Atzler C, Ege A, Knorr T, Koszinowski U, Kolanus W, Haas J (2001) Signaling by human herpesvirus 8 kaposin A through direct membrane recruitment of cytohesin-1. Mol Cells 7:833–843
Ko YC, Tsai WH, Wang PW, Wu IL, Lin SY, Chen YL, Chen JY, Lin SF (2012) Suppressive regulation of KSHV RTA with O-GlcNAcylation. J Biomed Sci 19:12
Koyano S, Mar EC, Stamey FR, Inoue N (2003) Glycoproteins M and N of human herpesvirus 8 form a complex and inhibit cell fusion. J Gen Virol 84:1485–1491
Krishnan HH, Sharma-Walia N, Zeng L, Gao SJ, Chandran B (2005) Envelope glycoprotein gB of Kaposi’s sarcoma-associated herpesvirus is essential for egress from infected cells. J Virol 79:10952–10967
Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD (2002) Protein interactions targeting the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus to cell chromosomes. J Virol 76:11596–11604
Krueger A, Baumann S, Krammer PH, Kirchhoff S (2001) FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol 21:8247–8254
Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D (2002) De novo infection and serial transmission of Kaposi’s Sarcoma-associated herpesvirus in cultured endothelial cells. J Virol 76:2440–2448
Lai IY, Farrell PJ, Kellam P (2011) X-box binding protein 1 induces the expression of the lytic cycle transactivator of Kaposi’s sarcoma-associated herpesvirus but not Epstein–Barr virus in co-infected primary effusion lymphoma. J Gen Virol 92:421–431
Laman H, Coverley D, Krude T, Laskey R, Jones N (2001) Viral cyclin–cyclin-dependent kinase 6 complexes initiate nuclear DNA replication. Mol Cell Biol 21:624–635
Lan K, Kuppers DA, Verma SC, Robertson ES (2004) Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J Virol 78:6585–6594
Lan K, Kuppers DA, Robertson ES (2005a) Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J Virol 79:3468–3478
Lan K, Kuppers DA, Verma SC, Sharma N, Murakami M, Robertson ES (2005b) Induction of Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J Virol 79:7453–7465
Lan K, Murakami M, Choudhuri T, Kuppers DA, Robertson ES (2006) Intracellular-activated Notch1 can reactivate Kaposi’s sarcoma-associated herpesvirus from latency. Virology 351:393–403
Lavreys L, Chohan B, Ashley R, Richardson BA, Corey L, Mandaliya K, Ndinya-Achola JO, Kreiss JK (2003) Human herpesvirus 8: seroprevalence and correlates in prostitutes in Mombasa, Kenya. J Infect Dis 187:359–363
Lee HR, Lee S, Chaudhary PM, Gill P, Jung JU (2010) Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Future Microbiol 5:1349–1365
Lee HR, Brulois K, Wong L, Jung JU (2012) Modulation of immune system by Kaposi’s Sarcoma-associated herpesvirus: lessons from viral evasion strategies. Front Microbiol 3:44
Lee HR, Choi UY, Hwang SW, Kim S, Jung JU (2016a) Viral inhibition of PRR-mediated innate immune response: learning from KSHV evasion strategies. Mol Cells 39:777–782
Lee HR, Mitra J, Lee S, Gao SJ, Oh TK, Kim MH, Ha T, Jung JU (2016b) Kaposi’s Sarcoma-associated herpesvirus viral interferon regulatory factor 4 (vIRF4) perturbs the G1-S cell cycle progression via deregulation of the cyclin D1 gene. J Virol 90:1139–1143
Lee MS, Yuan H, Jeon H, Zhu Y, Yoo S, Shi S, Krueger B, Renne R, Lu C, Jung JU, Gao SJ (2016c) Human mesenchymal stem cells of diverse origins support persistent infection with Kaposi’s sarcoma-associated herpesvirus and manifest distinct angiogenic, invasive, and transforming phenotypes. MBio 7:e02109–e02115
Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ (2010) Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol 12:193–199
Lennette ET, Blackbourn DJ, Levy JA (1996) Antibodies to human herpesvirus type 8 in the general population and in Kaposi’s sarcoma patients. Lancet 348:858–861
Li J, Huang Y, Cockerell CJ, Friedman-Kien AE (1996) Localization of human herpes-like virus type 8 in vascular endothelial cells and perivascular spindle-shaped cells of Kaposi’s Sarcoma lesions by in situ hybridization. Am J Pathol 148:1741–1748
Li M, Lee H, Yoon DW, Albrecht JC, Fleckenstein B, Neipel F, Jung JU (1997) Kaposi’s sarcoma-associated herpesvirus encodes a functional cyclin. J Virol 71:1984–1991
Li FY, Jeffrey PD, Yu JW, Shi Y (2006) Crystal structure of a viral FLIP: insights into FLIP-mediated inhibition of death receptor signaling. J Biol Chem 281:2960–2968
Li Q, Zhou F, Ye F, Gao SJ (2008) Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 379:234–244
Li W, Avey D, Fu B, Wu JJ, Ma S, Liu X, Zhu F (2016a) Kaposi’s sarcoma-associated herpesvirus inhibitor of cGAS (KicGAS), encoded by ORF52, is an abundant tegument protein and is required for production of infectious progeny viruses. J Virol 90:5329–5342
Li W, Jia XM, Shen C, Zhang M, Xu J, Shang Y, Zhu K, Hu M, Yan Q, Qin D, Lee M, Zhu J, Lu H, Krueger BJ, Renne R, Gao SJ, Lu C (2016b) A KSHV microRNA enhances viral latency and induces angiogenesis by targeting GRK2 to activate the CXCR164/AKT pathway. Oncotarget 7:32286–32305
Li W, Yan Q, Ding X, Shen C, Hu M, Zhu Y, Qin D, Lu H, Krueger BJ, Renne R, Gao SJ, Lu C (2016c) The SH3BGR/STAT3 pathway regulates cell migration and angiogenesis induced by a gammaherpesvirus MicroRNA. PLoS Pathog 12:e1005605
Li W, Hu M, Wang C, Lu H, Chen F, Xu J, Shang Y, Wang F, Qin J, Yan Q, Krueger BJ, Renne R, Gao SJ, Lu C (2017) A viral microRNA downregulates metastasis suppressor CD82 and induces cell invasion and angiogenesis by activating the c-Met signaling. Oncogene 36:5407–5420
Li Y, Zhong C, Liu D, Yu W, Chen W, Wang Y, Shi S, Yuan Y (2018) Evidence for Kaposi sarcoma originating from Mesenchymal stem cell through KSHV-induced Mesenchymal-to-endothelial transition. Cancer Res 78:230–245
Liang D, Gao Y, Lin X, He Z, Zhao Q, Deng Q, Lan K (2011) A human herpesvirus miRNA attenuates interferon signaling and contributes to maintenance of viral latency by targeting IKK epsilon. Cell Res 21:793–806
Liao W, Tang Y, Lin SF, Kung HJ, Giam CZ (2003) K-bZIP of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-Mediated transactivation. J Virol 77:3809–3815
Lim C, Sohn H, Gwack Y, Choe J (2000) Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) binds ATF4/CREB2 and inhibits its transcriptional activation activity. J Gen Virol 81:2645–2652
Lim C, Sohn H, Lee D, Gwack Y, Choe J (2002) Functional dissection of latency-associated nuclear antigen 1 of Kaposi’s sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J Virol 76:10320–10331
Lin C, Li H, Wang Y, Zhu FX, Kudchodkar S, Yuan Y (2003) Kaposi’s Sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: identification of the ori-Lyt and association of K8 bZip protein with the origin. J Virol 77:5578–5588
Lin X, Liang D, He Z, Deng Q, Robertson E, Lan K (2011) miR-K12-7-5p encoded by Kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS ONE 6:e16224
Lin X, Li X, Liang D, Lan K (2012) MicroRNAs and unusual small RNAs discovered in Kaposi’s sarcoma-associated herpesvirus virions. J Virol 86:12717–12730
Lisco A, Barbierato M, Fiore JR, Gasperini P, Favia A, Volpe A, Chironna M, Pastore G, Chieco-Bianchi L, Calabro ML (2006) Pregnancy and human herpesvirus 8 reactivation in human immunodeficiency virus type 1-infected women. J Clin Microbiol 44:3863–3871
Liu Z, Fang Q, Zhou S, Minhas V, Wood C, He N, Zhang T (2017) Seroprevalence of Kaposi’s sarcoma-associated herpesvirus among HIV-infected Uygurs in Xinjiang, China. J Med Virol 89:1629–1635
Liu D, Wang Y, Yuan Y (2018) Kaposi’s sarcoma-associated herpesvirus K8 is an RNA binding protein that regulates viral DNA replication in coordination with a noncoding RNA. J Virol 92:e02177-17
Lock MJ, Thorley N, Teo J, Emery VC (2002) Azidodeoxythymidine and didehydrodeoxythymidine as inhibitors and substrates of the human herpesvirus 8 thymidine kinase. J Antimicrob Chemother 49:359–366
Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, Lieberman PM (2003) Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J Virol 77:11425–11435
Lu F, Day L, Gao SJ, Lieberman PM (2006) Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J Virol 80:5273–5282
Lu CC, Li Z, Chu CY, Feng J, Feng J, Sun R, Rana TM (2010) MicroRNAs encoded by Kaposi’s sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep 11:784–790
Lukac DM, Kirshner JR, Ganem D (1999a) Transcriptional activation by the product of open reading frame 50 of Kaposi’s Sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J Virol 73:9348–9361
Lukac DM, Kirshner JR, Ganem D (1999b) Transcriptional activation by the product of open reading frame 50 of Kaposi’s sarcoma-associated herpesvirus Is required for lytic viral reactivation in B cells. J Virol 73:9348–9361
Lukac DM, Garibyan L, Kirshner JR, Palmeri D, Ganem D (2001) DNA binding by Kaposi’s sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J Virol 75:6786–6799
Majerciak V, Zheng ZM (2009) Kaposi’s sarcoma-associated herpesvirus ORF57 in viral RNA processing. Front Biosci 14:1516–1528
Majerciak V, Zheng ZM (2015) KSHV ORF57, a protein of many faces. Viruses 7:604–633
Majerciak V, Zheng ZM (2016) Alternative RNA splicing of KSHV ORF57 produces two different RNA isoforms. Virology 488:81–87
Majerciak V, Yamanegi K, Nie SH, Zheng ZM (2006a) Structural and functional analyses of Kaposi sarcoma-associated herpesvirus ORF57 nuclear localization signals in living cells. J Biol Chem 281:28365–28378
Majerciak V, Yamanegi K, Zheng ZM (2006b) Gene structure and expression of Kaposi’s sarcoma-associated herpesvirus ORF56, ORF57, ORF58, and ORF59. J Virol 80:11968–11981
Majerciak V, Pripuzova N, McCoy JP, Gao SJ, Zheng ZM (2007) Targeted disruption of Kaposi’s sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, K8alpha, and K8.1 and the production of infectious virus. J Virol 81:1062–1071
Majerciak V, Yamanegi K, Allemand E, Kruhlak M, Krainer AR, Zheng ZM (2008) Kaposi’s sarcoma-associated herpesvirus ORF57 functions as a viral splicing factor and promotes expression of intron-containing viral lytic genes in spliceosome-mediated RNA splicing. J Virol 82:2792–2801
Majerciak V, Uranishi H, Kruhlak M, Pilkington GR, Massimelli MJ, Bear J, Pavlakis GN, Felber BK, Zheng ZM (2011) Kaposi’s sarcoma-associated herpesvirus ORF57 interacts with cellular RNA export cofactors RBM15 and OTT3 to promote expression of viral ORF59. J Virol 85:1528–1540
Majerciak V, Ni T, Yang W, Meng B, Zhu J, Zheng ZM (2013) A viral genome landscape of RNA polyadenylation from KSHV latent to lytic infection. PLoS Pathog 9:e1003749
Majerciak V, Lu M, Li X, Zheng ZM (2014) Attenuation of the suppressive activity of cellular splicing factor SRSF3 by Kaposi sarcoma-associated herpesvirus ORF57 protein is required for RNA splicing. RNA 20:1747–1758
Majerciak V, Pripuzova N, Chan C, Temkin N, Specht SI, Zheng ZM (2015) Stability of structured Kaposi’s sarcoma-associated herpesvirus ORF57 protein is regulated by protein phosphorylation and homodimerization. J Virol 89:3256–3274
Malik P, Blackbourn DJ, Clements JB (2004) The evolutionarily conserved Kaposi’s sarcoma-associated herpesvirus ORF57 protein interacts with REF protein and acts as an RNA export factor. J Biol Chem 279:33001–33011
Malope BI, MacPhail P, Mbisa G, MacPhail C, Stein L, Ratshikhopha EM, Ndhlovu L, Sitas F, Whitby D (2008) No evidence of sexual transmission of Kaposi’s sarcoma herpes virus in a heterosexual South African population. AIDS 22:519–526
Mantina H, Kankasa C, Klaskala W, Brayfield B, Campbell J, Du Q, Bhat G, Kasolo F, Mitchell C, Wood C (2001) Vertical transmission of Kaposi’s sarcoma-associated herpesvirus. Int J Cancer 94:749–752
Martin JN, Ganem D, Osmond DH, Page-Shafer KA, Macrae D, Kedes DH (1998) Sexual transmission and the natural history of human herpesvirus 8 infection. N Engl J Med 338:948–954
Martinez FP, Tang Q (2012) Leucine zipper domain is required for Kaposi sarcoma-associated herpesvirus (KSHV) K-bZIP protein to interact with histone deacetylase and is important for KSHV replication. J Biol Chem 287:15622–15634
Massimelli MJ, Kang JG, Majerciak V, Le SY, Liewehr DJ, Steinberg SM, Zheng ZM (2011) Stability of a long noncoding viral RNA depends on a 9-nt core element at the RNA 5′ end to interact with viral ORF57 and cellular PABPC1. Int J Biol Sci 7:1145–1160
Massimelli MJ, Majerciak V, Kruhlak M, Zheng ZM (2013) Interplay between polyadenylate-binding protein 1 and Kaposi’s sarcoma-associated herpesvirus ORF57 in accumulation of polyadenylated nuclear RNA, a viral long noncoding RNA. J Virol 87:243–256
Massimelli MJ, Majerciak V, Kang JG, Liewehr DJ, Steinberg SM, Zheng ZM (2015) Multiple regions of Kaposi’s sarcoma-associated herpesvirus ORF59 RNA are required for its expression mediated by viral ORF57 and cellular RBM15. Viruses 7:496–510
Matta H, Sun Q, Moses G, Chaudhary PM (2003) Molecular genetic analysis of human herpes virus 8-encoded viral FLICE inhibitory protein-induced NF-kappaB activation. J Biol Chem 278:52406–52411
Matta H, Mazzacurati L, Schamus S, Yang T, Sun Q, Chaudhary PM (2007) Kaposi’s sarcoma-associated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IkappaB kinase complex to selectively activate NF-kappaB without JNK activation. J Biol Chem 282:24858–24865
Matta H, Gopalakrishnan R, Graham C, Tolani B, Khanna A, Yi H, Suo Y, Chaudhary PM (2012) Kaposi’s sarcoma associated herpesvirus encoded viral FLICE inhibitory protein K13 activates NF-kappaB pathway independent of TRAF6, TAK1 and LUBAC. PLoS ONE 7:e36601
McAllister SC, Hansen SG, Ruhl RA, Raggo CM, DeFilippis VR, Greenspan D, Früh K, Moses AV (2004) Kaposi sarcoma–associated herpesvirus (KSHV) induces heme oxygenase-1 expression and activity inKSHV-infected endothelial cells. Blood 103:3465–3473
McCormick C, Ganem D (2005) The Kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307:739–741
Meade-Tollin LC, Way D, Witte MH (1999) Expression of multiple matrix metalloproteinases and urokinase type plasminogen activator in cultured Kaposi sarcoma cells. Acta Histochem 101:305–316
Melbye M, Cook PM, Hjalgrim H, Begtrup K, Simpson GR, Biggar RJ, Ebbesen P, Schulz TF (1998) Risk factors for Kaposi’s-sarcoma-associated herpesvirus (KSHV/HHV-8) seropositivity in a cohort of homosexual men, 1981–1996. Int J Cancer 77:543–548
Miller G, Heston L, Grogan E, Gradoville L, Rigsby M, Sun R, Shedd D, Kushnaryov VM, Grossberg S, Chang Y (1997) Selective switch between latency and lytic replication of Kaposi’s sarcoma herpesvirus and Epstein–Barr virus in dually infected body cavity lymphoma cells. J Virol 71:314–324
Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L (2007) Lytic cycle switches of oncogenic human gammaherpesviruses 1. Adv Cancer Res 97:81–109
Minhas V, Wood C (2014) Epidemiology and transmission of Kaposi’s sarcoma-associated herpesvirus. Viruses 6:4178–4194
Molden J, Chang Y, You Y, Moore PS, Goldsmith MA (1997) A Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J Biol Chem 272:19625–19631
Moldovan GL, Pfander B, Jentsch S (2007) PCNA, the maestro of the replication fork. Cell 129:665–679
Mollica L, Bessa LM, Hanoulle X, Jensen MR, Blackledge M, Schneider R (2016) binding mechanisms of intrinsically disordered proteins: theory, simulation, and experiment. Front Mol Biosci 3:52
Moody R, Zhu Y, Huang Y, Cui X, Jones T, Bedolla R, Lei X, Bai Z, Gao SJ (2013) KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog 9:e1003857
Moore PS, Chang Y (2003) Kaposi’s sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu Rev Microbiol 57:609–639
Muralidhar S, Pumfery AM, Hassani M, Reza Sadaie M, Azumi N, Kishishita M, Brady JN, Doniger J, Medveczky P, Rosenthal LJ (1998) Identification of Kaposin (open reading frame K12) as a human Herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) transforming gene. J Virol 72:4980–4988
Myoung J, Ganem D (2011a) Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21
Myoung J, Ganem D (2011b) Infection of primary human tonsillar lymphoid cells by KSHV reveals frequent but abortive infection of T cells. Virology 413:1–11
Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O (2009) Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5:376–385
Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU (2003) Global changes in Kaposi’s sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220
Naranatt PP, Akula SM, Chandran B (2002) Characterization of gamma2-human herpesvirus-8 glycoproteins gH and gL. Arch Virol 147:1349–1370
Neipel F, Albrecht JC, Fleckenstein B (1997) Cell-homologous genes in the Kaposi’s sarcoma-associated Rhadinovirus Human Herpesvirus 8: determinants of its pathogenicity? J Virol 71:4187–4192
Nekorchuk M, Han Z, Hsieh TT, Swaminathan S (2007) Kaposi’s sarcoma-associated herpesvirus ORF57 protein enhances mRNA accumulation independently of effects on nuclear RNA export. J Virol 81:9990–9998
O’Byrne KJ, Dalgleish AG, Browning MJ, Steward WP, Harris AL (2000) The relationship between angiogenesis and the immune response in carcinogenesis and the progression of malignant disease. Eur J Cancer 36:151–169
Ohlsson R, Renkawitz R, Labanenkov V (2001) CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet 17:520–527
Ohsaki E, Ueda K (2012) Kaposi’s sarcoma-associated herpesvirus genome replication, partitioning, and maintenance in latency. Front Microbiol 3:7
Ohsaki E, Ueda K, Sakakibara S, Do E, Yada K, Yamanishi K (2004) Poly(ADP-ribose) polymerase 1 binds to Kaposi’s sarcoma-associated herpesvirus (KSHV) terminal repeat sequence and modulates KSHV replication in latency. J Virol 78:9936–9946
Okroj M, Tedeschi R, Mancuso R, Brambilla L, Tourlaki A, Dillner J, Blom AM (2011) Prevalence of antibodies against Kaposi’s sarcoma associated herpes virus (KSHV) complement inhibitory protein (KCP) in KSHV-related diseases and their correlation with clinical parameters. Vaccine 29:1129–1134
Orenstein JM (2008) Ultrastructure of Kaposi Sarcoma. Ultrastruct Pathol 32:211–220
Ottinger M, Christalla T, Nathan K, Brinkmann MM, Viejo-Borbolla A, Schulz TF (2006) Kaposi’s sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J Virol 80:10772–10786
Patel V, Dahlroth SL, Rajakannan V, Ho HT, Cornvik T, Nordlund P (2015) Structure of the C-terminal domain of the multifunctional ICP27 protein from herpes simplex virus 1. J Virol 89:8828–8839
Paul AG, Chandran B, Sharma-Walia N (2013) Cyclooxygenase-2-prostaglandin E2-eicosanoid receptor inflammatory axis: a key player in Kaposi’s sarcoma-associated herpes virus associated malignancies. Transl Res 162:77–92
Pearce M, Matsumura S, Wilson AC (2005) Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi’s sarcoma-associated herpesvirus originate from a common promoter. J Virol 79:14457–14464
Persson LM, Wilson AC (2010) Wide-scale use of Notch signaling factor CSL/RBP-Jkappa in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J Virol 84:1334–1347
Pertel PE (2002) Human herpesvirus 8 glycoprotein B (gB), gH, and gL can mediate cell fusion. J Virol 76:4390–4400
Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grässer FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T (2005) Identification of microRNAs of the herpesvirus family. Nat Methods 2:269–276
Picchio GR, Sabbe RE, Gulizia RJ, Mcgrath M, Herndier BG, Moiser DE (1997) The KSHV/HHV8-infected BCBL-1 lymphoma line causes tumors in SCID Mice but fails to transmit virus to a human peripheral blood mononuclear cell graft. J Virol Methods 238:22–29
Pilkington GR, Majerciak V, Bear J, Uranishi H, Zheng ZM, Felber BK (2012) Kaposi’s sarcoma-associated herpesvirus ORF57 is not a bona fide export factor. J Virol 86:13089–13094
Purushothaman P, McDowell ME, McGuinness J, Salas R, Rumjahn SM, Verma SC (2012) Kaposi’s sarcoma-associated herpesvirus-encoded LANA recruits topoisomerase IIbeta for latent DNA replication of the terminal repeats. J Virol 86:9983–9994
Purushothaman P, Thakker S, Verma SC (2015) Transcriptome analysis of Kaposi’s sarcoma-associated herpesvirus during de novo primary infection of human B and endothelial cells. J Virol 89:3093–3111
Purushothaman P, Uppal T, Sarkar R, Verma SC (2016) KSHV-mediated angiogenesis in tumor progression. Viruses 8:E198
Qin J, Li W, Gao SJ, Lu C (2017) KSHV microRNAs: tricks of the devil. Trends Microbiol 25:648–661
Quinlivan EB, Zhang C, Stewart PW, Komoltri C, Davis MG, Wehbie RS (2002) Elevated virus loads of Kaposi’s sarcoma–associated human herpesvirus 8 predict Kaposi’s sarcoma disease progression, but elevated levels of human immunodeficiency virus type 1 do not. J Infect Dis 185:1736–1744
Radkov SA, Kellam R, Boshoff C (2000) The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma–E2F pathway and with the oncogene Hras transforms primary rat cells. Nat Med 6:1121–1127
Radu O, Pantanowitz L (2013) Kaposi Sarcoma. Arch Pathol Lab Med 137:289–294
Ramalingam D, Happel C, Ziegelbauer JM (2015) Kaposi’s sarcoma-associated herpesvirus microRNAs repress breakpoint cluster region protein expression, enhance Rac1 activity, and increase in vitro angiogenesis. J Virol 89:4249–4261
Rappocciolo G, Jenkins FJ, Hensler HR, Piazza P, Jais M, Borowski L, Watkins SC, Rinaldo CR (2006) DC-SIGN is a receptor for human herpesvirus 8 on dendritic cells and macrophages. J Immunol 176:1741–1749
Renne R, Lagunoff M, Zhong W, Ganem D (1996a) The size and conformation of Kaposi’s Sarcoma-associated Herpesvirus (Human Herpesvirus 8) DNA in infected cells and virions. J Virol 70:8151–8154
Renne R, Zhong W, Herndier B, Mcgrath M, Abbey N, Kedes D, Ganem D (1996b) Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2:342–346
Renne R, Dittmer D, Kedes D, Schmidt K, Desrosiers RC, Luciw PA, Ganem D (2004) Experimental transmission of Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) to SIV-positive and SIV-negative rhesus macaques. J Med Primatol 33:1–9
Revilla-López J, Mendoza-Fabián R, Anampa-Guzmán A, Rosales-Zúñiga J, Chalco-Huamán J (2015) Sarcoma de Kaposi endemico en un paciente VIH negativo. Rev Peru Med Exp Salud Publica 32:808–812
Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y (2001) Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J Virol 75:429–438
Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore P (1996) Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci USA 93:14862–14867
Sadagopan S, Sharma-Walia N, Veettil MV, Bottero V, Levine R, Vart RJ, Chandran B (2009) Kaposi’s sarcoma-associated herpesvirus upregulates angiogenin during infection of human dermal microvascular endothelial cells, which induces 45S rRNA synthesis, antiapoptosis, cell proliferation, migration, and angiogenesis. J Virol 83:3342–3364
Sadler R, Wu L, Forghani B, Renne R, Zhong W, Herndier B, Ganem D (1999) A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J Virol 73:5722–5730
Sakakibara S, Tosato G (2009) Regulation of angiogenesis in malignancies associated with Epstein–Barr virus and Kaposi’s sarcoma-associated herpes virus. Future Microbiol 4:903–917
Sarek G, Jarviluoma A, Ojala PM (2006) KSHVviral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood 107:725–732
Sarek G, Jarviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M, Ojala PM (2010) Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog 6:e1000818
Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS (1999) Transcription mapping of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J Virol 72:1005–1012
Sathish N, Wang X, Yuan Y (2012) Tegument Proteins of Kaposi’s sarcoma-associated herpesvirus and related gamma-herpesviruses. Front Microbiol 3:98
Saveliev AK, Zhu FX, Yuan Y (2002) Transcription mapping and expression patterns of genes in the major immediate-early region of Kaposi’s sarcoma-associated herpesvirus. Virology 299:301–314
Schaefke B, Sun W, Li YS, Fang L, Chen W (2018) The evolution of posttranscriptional regulation. Wiley Interdiscip Rev RNA 9:e1485
Shamay M, Liu J, Li R, Liao G, Shen L, Greenway M, Hu S, Zhu J, Xie Z, Ambinder RF, Qian J, Zhu H, Hayward SD (2012) A protein array screen for Kaposi’s sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J Virol 86:5179–5191
Sharma NR, Majerciak V, Kruhlak MJ, Zheng ZM (2017) KSHV inhibits stress granule formation by viral ORF57 blocking PKR activation. PLoS Pathog 13:e1006677
Sharma-Walia N, Paul AG, Bottero V, Sadagopan S, Veettil MV, Kerur N, Chandran B (2010) Kaposi’s sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog 6:e1000777
Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, Du MQ, Boshoff C (2002) K15 protein of Kaposi’s sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J Virol 76:802–816
Shin YC, Nakamura H, Liang X, Feng P, Chang H, Kowalik TF, Jung JU (2006) Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J Virol 80:2257–2266
Shin HJ, DeCotiis J, Giron M, Palmeri D, Lukac DM (2014) Histone deacetylase classes I and II regulate Kaposi’s sarcoma-associated herpesvirus reactivation. J Virol 88:1281–1292
Simpson GR, Schulz TF, Whitby D, Cook PM, Boshoff C, Rainbow L, Howard MR, Gao S-J, Bohenzky RA, Simmonds P, Lee C, de Ruiter A, Hatzakis A, Tedder RS, Weller IVD, Weiss RA, Moore PS (1996) Prevalence of Kaposi’s sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet 348:1133–1138
Singh VV, Dutta D, Ansari MA, Dutta S, Chandran B (2014) Kaposi’s sarcoma-associated herpesvirus induces the ATM and H2AX DNA damage response early during de novo infection of primary endothelial cells, which play roles in latency establishment. J Virol 88:2821–2834
Skalsky RL, Hu J, Renne R (2007) Analysis of viral cis elements conferring Kaposi’s sarcoma-associated herpesvirus episome partitioning and maintenance. J Virol 81:9825–9837
Smith NA, Sabin CA, Gopal R, Bourboulia D, Labbet W, Boshoff C, Barlow D, Band B, Peters BS, de Ruiter A, Brown DWG, Weiss RA, Best JM, Whitby D (1999) Serologic evidence of Human Herpesvirus 8 transmission by homosexual but not heterosexual sex. J Infect Dis 180:600–606
Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem P, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F (1995) Kaposi’s Sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86:1276–1280
Speck SH, Ganem D (2010) Viral latency and its regulation: lessons from the gammaherpesviruses. Cell Host Microbe 8:100–115
Spiller OB, Robinson M, O’Donnell E, Milligan S, Morgan BP, Davison AJ, Blackbourn DJ (2003) Complement regulation by Kaposi’s sarcoma-associated herpesvirus ORF4 protein. J Virol 77:592–599
Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT (1997) Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol 71:715–719
Stedman W, Deng Z, Lu F, Lieberman PM (2004) ORC, MCM, and histone hyperacetylation at the Kaposi’s sarcoma-associated herpesvirus latent replication origin. J Virol 78:12566–12575
Sturzl M, Blasig C, Schreier A, Neipel F, Hohenadl C, Cornali E, Ascherl G, Esser S, Brockmeyer NH, Ekman M, Kaaya EE, Tschachler E, Biberfeld P (1997) Expression of HHV-8 latency-associated T0.7 RNA in spindle cells and endothelial cells of AIDS-associated, classical and African Kaposi’s Sarcoma. Int J Cancer 72:68–71
Sturzl M, Hohenadl C, Zietz C, Castanos-Velez E, Wunderlich A, Ascherl G, Biberfeld P, Monini P, Browning PJ, Ensoli B (1999) Expression of K13/v-FLIP gene of human herpesvirus 8 and apoptosis in Kaposi’s sarcoma spindle cells. J Natl Cancer Inst 91:1725–1733
Sullivan CS (2007) High conservation of Kaposi sarcoma–associated herpesvirus microRNAs implies important function. J Infect Dis 195:618–620
Sun R, Lin S, Staskus KA, Gradoville L, Grogan E, Haase A, Miller G (1999) Kinetics of Kaposi’s Sarcoma-associated herpesvirus gene expression. J Virol 73:2232–2242
Sun Q, Tsurimoto T, Juillard F, Li L, Li SJ, De León Vázquez E, Chen S, Kaye K (2014a) Kaposi’s sarcoma-associated herpesvirus LANA recruits the DNA polymerase clamp loader to mediate efficient replication and virus persistence. Proc Natl Acad Sci USA 111:11816–11821
Sun R, Liang D, Gao Y, Lan K (2014b) Kaposi’s sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J Virol 88:7331–7344
Sun Z, Jha HC, Robertson ES (2015) Bub1 in complex with LANA recruits PCNA to regulate kaposi’s sarcoma-associated herpesvirus latent replication and DNA translesion synthesis. J Virol 89:10206–10218
Tan B, Gao SJ (2018) RNA epitranscriptomics: regulation of infection of RNA and DNA viruses by N(6) -methyladenosine (m(6) A). Rev Med Virol 28:e1983
Tan B, Liu H, Zhang S, da Silva SR, Zhang L, Meng J, Cui X, Yuan H, Sorel O, Zhang S-W, Huang Y, Gao SJ (2017) Viral and cellular N6-methyladenosine and N6,2′-O-dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat Microbiol 3:108–120
Tang S, Zheng ZM (2002) Kaposi’s sarcoma-associated herpesvirus K8 exon 3 contains three 5′-splice sites and harbors a K8.1 transcription start site. J Biol Chem 277:14547–14556
Tang Q, Qin D, Lv Z, Zhu X, Ma X, Yan Q, Zeng Y, Guo Y, Feng N, Lu C (2012) Herpes simplex virus type 2 triggers reactivation of Kaposi’s sarcoma-associated herpesvirus from latency and collaborates with HIV-1 Tat. PLoS ONE 7:e31652
Tomkowicz B, Singh SP, Cartas M, Srinivasan A (2002) Human herpesvirus-8 encoded Kaposin: subcellular localization using immunofluorescence and biochemical approaches. DNA Cell Biol 21:151–162
Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU (2010) Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog 6:e1001013
Toth Z, Brulois K, Jung JU (2013a) The chromatin landscape of Kaposi’s sarcoma-associated herpesvirus. Viruses 5:1346–1373
Toth Z, Brulois K, Lee HR, Izumiya Y, Tepper C, Kung HJ, Jung JU (2013b) Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog 9:e1003813
Toth Z, Papp B, Brulois K, Choi YJ, Gao SJ, Jung JU (2016) LANA-mediated recruitment of host polycomb repressive complexes onto the KSHV genome during de novo infection. PLoS Pathog 12:e1005878
Tsai WH, Wang PW, Lin SY, Wu IL, Ko YC, Chen YL, Li M, Lin SF (2012) Ser-634 and Ser-636 of Kaposi’s sarcoma-associated herpesvirus RTA are involved in transactivation and are potential Cdk9 phosphorylation sites. Front Microbiol 3:60
Tsuyama T, Tada S, Watanabe S, Seki M, Enomoto T (2005) Licensing for DNA replication requires a strict sequential assembly of Cdc6 and Cdt1 onto chromatin in Xenopus egg extracts. Nucleic Acids Res 33:765–775
Tunnicliffe RB, Schacht M, Levy C, Jowitt TA, Sandri-Goldin RM, Golovanov AP (2015) The structure of the folded domain from the signature multifunctional protein ICP27 from herpes simplex virus-1 reveals an intertwined dimer. Sci Rep 5:11234
Tunnicliffe RB, Collins RF, Ruiz Nivia HD, Sandri-Goldin RM, Golovanov AP (2018) The ICP27 homology domain of the human cytomegalovirus protein UL69 adopts a dimer-of-dimers structure. MBio 9:e01112–e01118
Uldrick TS, Wang V, O’Mahony D, Aleman K, Wyvill KM, Marshall V, Steinberg SM, Pittaluga S, Maric I, Whitby D, Tosato G, Little RF, Yarchoan R (2010) An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin Infect Dis 51:350–358
Uppal T, Banerjee S, Sun Z, Verma SC, Robertson ES (2014) KSHV LANA-the master regulator of KSHV latency. Viruses 6:4961–4998
Van Bortle K, Corces VG (2013a) The role of chromatin insulators in nuclear architecture and genome function. Curr Opin Genet Dev 23:212–218
Van Bortle K, Corces VG (2013b) Spinning the web of cell fate. Cell 152:1213–1217
Van Dross R, Yao S, Asad S, Westlake G, Mays DJ, Barquero L, Duell S, Pietenpol JA, Browning PJ (2005) Constitutively active K-cyclin/cdk6 kinase in Kaposi sarcoma-associated herpesvirus-infected cells. J Natl Cancer Inst 97:656–666
Veettil MV, Bandyopadhyay C, Dutta D, Chandran B (2014) Interaction of KSHV with host cell surface receptors and cell entry. Viruses 6:4024–4046
Verma SC, Borah S, Robertson ES (2004) Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J Virol 78:10348–10359
Verma SC, Choudhuri T, Robertson ES (2007a) The minimal replicator element of the Kaposi’s sarcoma-associated herpesvirus terminal repeat supports replication in a semiconservative and cell-cycle-dependent manner. J Virol 81:3402–3413
Verma SC, Lan K, Choudhuri T, Cotter MA, Robertson ES (2007b) An autonomous replicating element within the KSHV genome. Cell Host Microbe 2:106–118
Verma SC, Lu J, Cai Q, Kosiyatrakul S, McDowell ME, Schildkraut CL, Robertson ES (2011) Single molecule analysis of replicated DNA reveals the usage of multiple KSHV genome regions for latent replication. PLoS Pathog 7:e1002365
Wang FZ, Akula SM, Pramod NP, Zeng L, Chandran B (2001) Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J Virol 75:7517–7527
Wang SE, Wu FY, Yu Y, Hayward GS (2003) CCAAT/enhancer-binding protein is induced during the early stages of Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J Virol 77:9590–9612
Wang X, He B, Zhang Z, Liu T, Wang H, Li X, Zhang Q, Lan K, Lu X, Wen H (2010) Human herpesvirus-8 in northwestern China: epidemiology and characterization among blood donors. Virol J 7:62
Wang Y, Sathish N, Hollow C, Yuan Y (2011) Functional characterization of Kaposi’s sarcoma-associated herpesvirus open reading frame K8 by bacterial artificial chromosome-based mutagenesis. J Virol 85:1943–1957
Wang L, Kang G, Kumar P, Lu W, Li Y, Zhou Y, Li Q, Wood C (2014) Humanized-BLT mouse model of Kaposi’s sarcoma-associated herpesvirus infection. Proc Natl Acad Sci USA 111:3146–3151
Wang F, Guo Y, Li W, Lu C, Yan Q (2018) Generation of a KSHV K13 deletion mutant for vFLIP function study. J Med Virol 90:753–760
Wen KW, Damania B (2010) Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett 289:140–150
Wen HJ, Minhas V, Wood C (2009) Identification and characterization of a new Kaposi’s sarcoma-associated herpesvirus replication and transcription activator (RTA)-responsive element involved in RTA-mediated transactivation. J Gen Virol 90:944–953
Whitby D, Howard MR, Tenant-Flowers M, Brink NS, Copas A, Boshoff C, Hatzioannou T, Suggett FEA, Aldam DM, Denton AS, Miller RF, Weller IVD, Weiss RA, Tedder RS, Schulz TF (1995) Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi’s sarcoma. Lancet 346:799–802
Woodard C, Shamay M, Liao G, Zhu J, Ng AN, Li R, Newman R, Rho HS, Hu J, Wan J, Qian J, Zhu H (2012) Phosphorylation of the chromatin binding domain of KSHV LANA. PLoS Pathog 8:e1002972
Wu J, Xu Y, Mo D, Huang P, Sun R, Huang L, Pan S, Xu J (2014) Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 promotes cell proliferation and migration by upregulating DNMT1 via STAT3 activation. PLoS ONE 9:e93478
Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, Konrad A, Neipel F, Sturzl M, Whitby D, Li H, Zhu F (2015) Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18:333–344
Xiao B, Verma SC, Cai Q, Kaul R, Lu J, Saha A, Robertson ES (2010) Bub1 and CENP-F can contribute to Kaposi’s sarcoma-associated herpesvirus genome persistence by targeting LANA to kinetochores. J Virol 84:9718–9732
Xie J, Ajibade AO, Ye F, Kuhne K, Gao SJ (2005) Reactivation of Kaposi’s sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 371:139–154
Yamanegi K, Tang S, Zheng ZM (2005) Kaposi’s sarcoma-associated herpesvirus K8beta is derived from a spliced intermediate of K8 pre-mRNA and antagonizes K8alpha (K-bZIP) to induce p21 and p53 and blocks K8alpha-CDK2 interaction. J Virol 79:14207–14221
Ye F (2017) RNA N6-adenosine methylation (m6A) steers epitranscriptomic control of herpesvirus replication. Inflamm Cell Signal 4:e1604
Ye F, Zhou F, Xie J, Kang T, Greene W, Kuhne K, Lei X, Li Q, Gao SJ (2008) Kaposi’s sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: a novel mechanism of virus control of latency. J Virol 82:4235–4249
Ye F, Lei X, Gao SJ (2011a) Mechanisms of Kaposi’s Sarcoma-associated herpesvirus latency and reactivation. Adv Virol 2011:193860
Ye F, Zhou F, Bedolla RG, Jones T, Lei X, Kang T, Guadalupe M, Gao SJ (2011b) Reactive oxygen species hydrogen peroxide mediates Kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog 7:e1002054
Ye F, Zhou F, Nithianantham S, Chandran B, Yu X, Weinberg A, Gao SJ (2013) Kaposi’s sarcoma-associated herpesvirus induces rapid release of angiopoietin-2 from endothelial cells. J Virol 87:6326–6335
Ye F, Chen ER, Nilsen TW (2017) Kaposi’s sarcoma-associated herpesvirus utilizes and manipulates RNA N6-Adenosine methylation to promote lytic replication. J Virol 91:e00466-00417
Yu F, Harada JN, Brown HJ, Deng H, Song MJ, Wu TT, Kato-Stankiewicz J, Nelson CG, Vieira J, Tamanoi F, Chanda SK, Sun R (2007) Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog 3:e44
Yuan F, Gao ZQ, Majerciak V, Bai L, Hu ML, Lin XX, Zheng ZM, Dong YH, Lan K (2018) The crystal structure of KSHV ORF57 reveals dimeric active sites important for protein stability and function. PLoS Pathog 14:e1007232
Zhang T, Liu Z, Wang J, Minhas V, Wood C, Clifford GM, He N, Franceschi S (2017) Seroprevalence of antibodies against Kaposi’s sarcoma-associated herpesvirus among HIV-negative people in China. Infect Agent Cancer 12:32
Zhao Q, Liang D, Sun R, Jia B, Xia T, Xiao H, Lan K (2015) Kaposi’s sarcoma-associated herpesvirus-encoded replication and transcription activator impairs innate immunity via ubiquitin-mediated degradation of myeloid differentiation factor 88. J Virol 89:415–427
Zheng ZM (2003) Split genes and their expression in Kaposi’s sarcoma-associated herpesvirus. Rev Med Virol 13:173–184
Zheng J, Yang Y, Cui M, Shu ZJ, Han LL, Liu ZQ, Wood C, Zhang T, Zeng Y (2017) Prevalence of Kaposi’s sarcoma-associated herpesvirus in Uygur and Han populations from the Urumqi and Kashgar regions of Xinjiang, China. Virol Sin 32:396–403
Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ (2002) Efficient infection by a recombinant Kaposi’s sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J Virol 76:6185–6196
Zhu FX, Cusano T, Yuan Y (1999) Identification of the immediate-early transcripts of Kaposi’s sarcoma-associated herpesvirus. J Virol 73:5556–5567
Zhu FX, Chong JM, Wu L, Yuan Y (2005) Virion proteins of Kaposi’s sarcoma-associated herpesvirus. J Virol 79:800–811
Ziegelbauer JM, Sullivan CS, Ganem D (2009) Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat Genet 41:130–134
Zoetweij JP, Moses A, Rinderknecht AS, Davis DA, Overwijk WW, Yarchoan R, Orenstein JM, Blauvelt A (2000) Targeted inhibition of calcineurin signaling blocks calcium-dependent reactivation of Kaposi sarcoma–associated herpesvirus. Blood 97:2374–2379
Acknowledgements
We apologize that many important references were omitted from this manuscript due to the page limitation. This work was supported by the National Key R&D Program of China (2016YFA0502100), the Natural Science Foundation for Distinguished Young Scholars (81425017), the National Institutes of Health awarded (7R01AI116442) to K.L, and the Intramural Research Program of NCI/NIH (1ZIASC010357) to ZMZ.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Animal and Human Rights Statement
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yan, L., Majerciak, V., Zheng, ZM. et al. Towards Better Understanding of KSHV Life Cycle: from Transcription and Posttranscriptional Regulations to Pathogenesis. Virol. Sin. 34, 135–161 (2019). https://doi.org/10.1007/s12250-019-00114-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12250-019-00114-3