
Abstract
During human immunodeficiency virus (HIV) infection, type I interferon (IFN-I) signaling induces an antiviral state that includes the production of restriction factors that inhibit virus replication, thereby limiting the infection. As seen in other viral infections, type I IFN can also increase systemic immune activation which, in HIV disease, is one of the strongest predictors of disease progression to acquired immune deficiency syndrome (AIDS) and non-AIDS morbidity and mortality. Moreover, IFN-I is associated with CD4 T cell depletion and attenuation of antigen-specific T cell responses. Therefore, therapeutic manipulation of IFN-I signaling to improve HIV disease outcome is a source of much interest and debate in the field. Recent studies have highlighted the importance of timing (acute vs. chronic infection) and have suggested that specific targeting of type I IFNs and their subtypes may help harness the beneficial roles of the IFN-I system while avoiding its deleterious activities.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
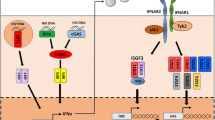
Type I interferons (IFN-I) are cytokines abundantly produced during various immunological conditions and are known to play a pivotal role in the control of several viral infections. In humans, the IFN-I family is composed of 13 IFNα subtypes, IFNβ, IFNδ, IFNε, IFNζ, IFNτ, IFNκ, and IFNω (Pestka et al.2004), of which IFNα and IFNβ are the most predominant and best characterized. All type I IFNs bind with various affinities to their common receptor IFNAR, engagement of which triggers downstream transcription of a vast array of IFN-simulated genes (ISGs). Complete absence of IFN-I signaling by deletion of IFNAR in mice has been shown to enhance mortality from vesicular somatic virus, West Nile virus and lymphocytic choriomeningitis virus (LCMV) (Muller et al.1994). With repercussions for both innate and adaptive immune responses, the overall immunological consequence of IFN-I signaling is highly contextual and may vary across viral infections, bacterial infections, auto-immune disorders, and so on. Here, we focus on viral infections, specifically human immunodeficiency virus (HIV) infection during which IFN-I signaling via ISG expression in immune cells can lead to an antiviral state that interferes with several aspects of virus replication (Yan and Chen 2012).
The Double Face of Type I IFNs
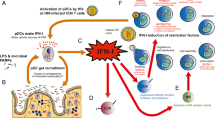
As early as a week after HIV infection, increased plasma levels of IFNα can be measured in infected individuals (Stacey et al.2009) and later on, elevated plasma levels of IFNα correlate with plasma HIV RNA and inversely with CD4 T cell count (Hardy et al.2013). Complementary data from animal studies have shown persistently higher ISG expression levels in simian immunodeficiency virus (SIV) infected rhesus macaques (Bosinger et al.2009; Jacquelin et al.2009; Sandler et al.2014). Importantly, IFN-I signaling resulting from SIV infection wanes during the transition from acute to chronic non-pathogenic infection in SIV natural hosts African green monkeys and sooty mangabeys, whereas a persistent response is associated with pathogenic infection and progression to acquired immune deficiency syndrome (AIDS) in experimental SIV infection of rhesus and pigtail macaques (Bosinger et al.2009; Jacquelin et al.2009; Harris et al.2010). Thus, these studies demonstrate a link between type I IFN signaling and pathogenic SIV infection.
Beneficial Effects of IFN-I Signaling
The notion that IFN-I signaling can induce a protective antiviral state in the infected host yet at the same time be linked to pathogenic disease progression may be counterintuitive at first. Reported mechanisms by which IFN-I signaling interferes with virus replication in HIV infection include, among others, expression of a specific set of ISGs referred to as restriction factors, that inhibit the virus at various stages of replication such as cell entry (TRIM5α), reverse transcription (SAMHD1 and APOBEC3), nuclear entry and integration (MX2), translation and assembly (SLFN 11 and GBP5) and budding (Tetherin) (Soper et al.2017). With over 300 known ISGs (Schoggins et al.2011; Liu et al.2012), the increasing number of reported restriction factors with functional effects observed in vitro and in vivo (Soper et al.2017; El-Diwany et al.2018) altogether reinforce the knowledge that IFN-I signaling is critical to the suppression of HIV during early stages of infection. Sandler demonstrated that protection from a systemic SIV infection upon intrarectal challenge was achieved in rhesus macaques by intramuscular administration of IFNα and was associated with increased upregulation of ISG including restriction factors (Sandler et al.2014). Likewise, upregulation of ISGs in vaginal cell suspensions was shown to associate with protection from simian/human immunodeficiency virus (SHIV) infection in rhesus macaques that had received vaginal IFNβ administration (Veazey et al.2016).
Detrimental Effects of IFN-I Signaling
Aside from restriction factors, other ISGs enhance pathogen associated molecular pattern (PAMP) detection and IFN signaling. For example, in response to viral PAMPs, IFN-I produced by plasmacytoid dendritic cells (pDCs) induces expression of the transcription factor IRF7 in neighboring cells which results in systemic expansion of IFN-I signaling. Similarly, other ISGs such as cGAMP and IFI16 all contribute to a persistent production of IFN-I (Bosinger and Utay 2015). The constant engagement of IFN-I signaling is thought to be at least in part responsible for the detrimental effects of IFN-I. Several studies have reported an association between IFN-I signaling and enhanced CD4 T cell depletion, potentially mediated through increased apoptosis, increase of CCR5-expressing HIV target cells or suppression of thymic output (Utay and Douek 2016). Moreover, studies on LCMV have shown that chronic IFN-I signaling is associated with immune suppression characterized by reduced antigen-specific T cell responses (Teijaro et al.2013; Wilson et al.2013), reduced T cell proliferation (Marshall et al.2011) and increased CD8 T cell exhaustion (Teijaro et al.2013; Wilson et al.2013). In HIV disease, type I interferon signaling is a major driver of immune activation, one of the strongest predictors of HIV disease progression to AIDS and non-AIDS morbidity and mortality (Hunt 2012). Collectively, these findings raised considerable interest in the potential therapeutic benefits of blocking IFN-I during HIV infection.
Lessons Learned from Manipulating IFN-I Signaling In Vivo
Enhancing IFN-I Signaling
Increased understanding of the opposing effects of type I IFN justifies the rationale for HIV/SIV studies aiming to enhance or block IFN-I signaling. In a considerable number of clinical studies (reviewed in Bosinger and Utay 2015), IFNα was administered subcutaneously, intramuscularly or orally to HIV infected persons not on antiretroviral therapy (ART). While some studies reported attenuation of CD4 T cell decline, lower HIV burden and opportunistic infections, others reported no significant improvements. These conflicting reports in conjunction with the efficacy of combinational ART underlined the impracticability of IFNα monotherapy for HIV treatment. A second wave of clinical studies investigated IFNα as supplement to ART. Data from over a dozen of studies showed no significant improvement of CD4 T cell recovery or clinical outcome, refuting a beneficial effect of IFNα as supplement to ART (Bosinger and Utay 2015). In a study by Massanella, pegylated interferon-α2a treatment of HIV infected persons on ART decreased absolute number of CD4 and CD8 T cells (Massanella et al.2010). More recently, studies largely conducted in animal models highlighted the importance of discriminating between three major HIV disease states: acute infection (where antiviral factors are upregulated), chronic ART-untreated infection (where both permanent low-level ISG expression and CD4 T cell depletion are maintained), and chronic ART-treated infection (where CD4 T cell recovery occurs and inflammation is at its lowest—albeit not normalized). Systemic administration of IFNα during rectal challenge transiently increased ISGs and prevented SIV acquisition in rhesus macaques (Sandler et al.2014) whereas administration of pegylated IFN-α2a to chronically SIV-infected ART-treated macaques upregulated ISGs with no effect on CD4 T cell depletion, immune activation or virus reservoir (Palesch et al.2018). Thus, in the acute stage of HIV infection, enhancing IFN-I signaling may be clinically beneficial but in the chronic stage there is to date no tangible evidence supporting such approach. In fact, the immunological consequences of a persisting immune activation would advocate for a suppression rather than enhancement of IFN-I signaling during chronic HIV infection.
Blocking IFN-I Signaling
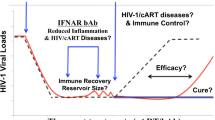
Blocking IFNAR during acute SIV infection in vivo increased SIV plasma viremia (Sandler et al.2014; Carnathan et al.2018), supporting the importance of IFN-I signaling for control of the infection during acute stage. In contrast, reduced inflammation and improved viral clearance was observed after blockade of IFN-I signaling with an anti-IFNAR antibody in murine LCMV infection (Teijaro et al.2013; Wilson et al.2013). Recently, two independent studies showed that administration of anti-IFNAR antibodies to ART-suppressed, HIV-infected humanized mice resulted in reduced immune activation and lowered reservoir size (Cheng et al.2017; Zhen et al.2017). The results of these mouse studies encouraged the idea that blocking IFN-I signaling during chronic HIV infection may help improve clinical outcome. In chronically SIV-infected rhesus macaques, systemic administration of a long half-life IFN-I antagonist significantly decreased ISGs with no impact on plasma virus load, immune activation or virus reservoir, irrespective of ART (Nganou-Makamdop et al.2018). With no obvious adverse effects, it remains to be determined what benefits will be gained from blocking IFN-I signaling during chronic HIV infection. Reduced T cell activation and virus reservoir achieved in humanized mice treated with anti-IFNAR blocking antibodies (Cheng et al.2017; Zhen et al.2017) was not observed in non-human primates treated with an IFNα antagonist (Nganou-Makamdop et al.2018). Recent studies demonstrated that virus control is mediated by IFNβ and T cell exhaustion by IFNα in murine LCMV infection (Ng et al.2015); and that in humanized HIV-infected mice, IFNβ or IFNα14 but not the commonly used IFNαa2 significantly suppressed HIV replication (Abraham et al.2016; Lavender et al.2016). Therefore, it is tempting to speculate that different members of the IFN-I family play different roles and manipulating specific type I IFNs might be needed to selectively target detrimental activities while maintaining beneficial ones.
Overall Outcome and Implication for HIV Infection
All HIV/SIV studies on type I IFNs undoubtedly concur that the effects of enhancing or suppressing IFN-I signaling are strongly influenced by the timing of treatment (acute or chronic infection). During acute infection, the antiviral effects of IFN-I outweigh the deleterious effects. IFN-I treatment has so far not been shown to match the efficacy reached with ART and would not be useful as monotherapy. However, ART alone does not resolve chronic immune activation (Fernandez et al.2011; Hunt 2012; Dunham et al.2014; Sereti et al.2017) and is not sufficient to completely purge the HIV reservoir (Henrich et al.2017), potentially because antiretroviral drugs do not reach sufficient levels in lymphoid tissues (Fletcher et al.2014) where HIV predominantly resides (Rothenberger et al.2015; Lorenzo-Redondo et al.2016). Because IFN-I readily penetrates tissues (Johns et al.1990), a legitimate question to address would be whether ART and IFN-I combination in acute HIV infection may impact the establishment or size of the virus reservoir. During chronic HIV, limiting IFN-I’s contribution to ongoing immune activation is thought to be a major target for clinical improvement. To date, this concept remains to be proven in experimental settings. Importantly, no study in non-human primate or humanized mice has so far shown a detrimental effect of blocking IFN-I signaling during chronic HIV/SIV infection; suggesting that type I IFNs may not be as critical for the control of the infection as they are in the acute phase.
A better understanding of the complex roles of IFN-I in HIV infection is likely to be achieved by addressing the following understudied questions:
-
Are all ISGs equally important to disease progression? In other words, do some of the many ISGs upregulated during acute infection or maintained during chronic infection associate with clinical outcome? Could these help predict disease course?
-
What are the major differences in IFN-I signaling or pathways induced by the various IFN-I types and could these help understand emerging data on their functional diversity in the control of HIV infection?
-
Can an optimal balance of maximal upregulation of restriction factors and minimal upregulation of immune activating ISGs be achieved? Can this help reduce the virus reservoir?
-
Can IFN-I (sub)type-specific targeting in chronic infection reduce systemic immune activation thereby ameliorating clinical outcome?
Noteworthy parallels exist between studies on HIV/SIV infection and studies on other viral infections. For instance, the paradoxical role of IFN-I in inducing antiviral responses and enhancing pathology has been described for influenza (Garcia-Sastre and Biron 2006; Davidson et al.2014) and SARS (Huang et al.2005). Taken together, data from several studies suggest that IFN-I signaling is essential to cytokine responses and recruitment of immune cells, which in turn can promote pathology. In influenza, administration of an agonist to the lipid metabolite sphingosine 1 phosphate receptor (S1P1R) expressed on pDCs was shown to suppress IFN-I signaling in mice and human cells and blunt proinflammatory responses during influenza infection in mice that showed improved survival (Teijaro et al.2011; Walsh et al.2011; Teijaro et al.2016). Thus, provided that optimal conditions are met, it may indeed be possible to manipulate IFN-I signaling to improve clinical outcome in HIV infection.
References
Abraham S, Choi JG, Ortega NM, Zhang J, Shankar P, Swamy NM (2016) Gene therapy with plasmids encoding IFN-beta or IFN-alpha14 confers long-term resistance to HIV-1 in humanized mice. Oncotarget 7:78412–78420
Bosinger SE, Utay NS (2015) Type I interferon: understanding its role in HIV pathogenesis and therapy. Curr HIV/AIDS Rep 12:41–53
Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, Francella N, Sidahmed A, Smith AJ, Cramer EM, Zeng M, Masopust D, Carlis JV, Ran L, Vanderford TH, Paiardini M, Isett RB, Baldwin DA, Else JG, Staprans SI, Silvestri G, Haase AT, Kelvin DJ (2009) Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Investig 119:3556–3572
Carnathan D, Lawson B, Yu J, Patel K, Billingsley JM, Tharp GK, Delmas OM, Dawoud R, Wilkinson P, Nicolette C, Cameron MJ, Sekaly RP, Bosinger SE, Silvestri G, Vanderford TH (2018) Reduced chronic lymphocyte activation following Interferon-alpha blockade in the acute phase of SIV infection in rhesus macaques. J Virol. https://doi.org/10.1128/jvi.01760-17
Cheng L, Ma J, Li J, Li D, Li G, Li F, Zhang Q, Yu H, Yasui F, Ye C, Tsao LC, Hu Z, Su L, Zhang L (2017) Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J Clin Investig 127:269–279
Davidson S, Crotta S, McCabe TM, Wack A (2014) Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun 5:3864
Dunham RM, Vujkovic-Cvijin I, Yukl SA, Broadhurst MJ, Loke P, Albright RG, Wong JK, Lederman MM, Somsouk M, Hunt PW, Martin JN, Deeks SG, McCune JM (2014) Discordance between peripheral and colonic markers of inflammation during suppressive ART. J Acquir Immune Defic Syndr 65:133–141
El-Diwany R, Soliman M, Sugawara S, Breitwieser F, Skaist A, Coggiano C, Sangal N, Chattergoon M, Bailey JR, Siliciano RF, Blankson JN, Ray SC, Wheelan SJ, Thomas DL, Balagopal A (2018) CMPK2 and BCL-G are associated with type 1 interferon-induced HIV restriction in humans. Sci Adv 4:eaat0843
Fernandez S, Tanaskovic S, Helbig K, Rajasuriar R, Kramski M, Murray JM, Beard M, Purcell D, Lewin SR, Price P, French MA (2011) CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J Infect Dis 204:1927–1935
Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, Beilman GJ, Khoruts A, Thorkelson A, Schmidt TE, Anderson J, Perkey K, Stevenson M, Perelson AS, Douek DC, Haase AT, Schacker TW (2014) Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci USA 111:2307–2312
Garcia-Sastre A, Biron CA (2006) Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882
Hardy GA, Sieg S, Rodriguez B, Anthony D, Asaad R, Jiang W, Mudd J, Schacker T, Funderburg NT, Pilch-Cooper HA, Debernardo R, Rabin RL, Lederman MM, Harding CV (2013) Interferon-alpha is the primary plasma type-I IFN in HIV-1 infection and correlates with immune activation and disease markers. PLoS ONE 8:e56527
Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, Silvestri G, Muller-Trutwin M, Vasile-Pandrea I, Apetrei C, Hirsch V, Lifson J, Brenchley JM, Estes JD (2010) Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol 84:7886–7891
Henrich TJ, Hatano H, Bacon O, Hogan LE, Rutishauser R, Hill A, Kearney MF, Anderson EM, Buchbinder SP, Cohen SE, Abdel-Mohsen M, Pohlmeyer CW, Fromentin R, Hoh R, Liu AY, McCune JM, Spindler J, Metcalf-Pate K, Hobbs KS, Thanh C, Gibson EA, Kuritzkes DR, Siliciano RF, Price RW, Richman DD, Chomont N, Siliciano JD, Mellors JW, Yukl SA, Blankson JN, Liegler T, Deeks SG (2017) HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: an observational study. PLoS Med 14:e1002417
Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, Lei HY (2005) An interferon-gamma-related cytokine storm in SARS patients. J Med Virol 75:185–194
Hunt PW (2012) HIV and inflammation: mechanisms and consequences. Curr HIV/AIDS Rep 9:139–147
Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, Dillies MA, Roques P, Butor C, Silvestri G, Giavedoni LD, Lebon P, Barre-Sinoussi F, Benecke A, Muller-Trutwin MC (2009) Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Investig 119:3544–3555
Johns TG, Kerry JA, Veitch BA, Mackay IR, Tutton PJ, Tymms MJ, Cheetham BF, Hertzog PJ, Linnane AW (1990) Pharmacokinetics, tissue distribution, and cell localization of [35S]methionine-labeled recombinant human and murine alpha interferons in mice. Cancer Res 50:4718–4723
Lavender KJ, Gibbert K, Peterson KE, Van Dis E, Francois S, Woods T, Messer RJ, Gawanbacht A, Muller JA, Munch J, Phillips K, Race B, Harper MS, Guo K, Lee EJ, Trilling M, Hengel H, Piehler J, Verheyen J, Wilson CC, Santiago ML, Hasenkrug KJ, Dittmer U (2016) Interferon alpha subtype-specific suppression of HIV-1 infection in vivo. J Virol 90:6001–6013
Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G (2012) Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci USA 109:4239–4244
Lorenzo-Redondo R, Fryer HR, Bedford T, Kim EY, Archer J, Pond SLK, Chung YS, Penugonda S, Chipman J, Fletcher CV, Schacker TW, Malim MH, Rambaut A, Haase AT, McLean AR, Wolinsky SM (2016) Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 530:51–56
Marshall HD, Urban SL, Welsh RM (2011) Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. J Virol 85:5929–5939
Massanella M, Tural C, Papagno L, Garcia E, Jou A, Bofill M, Autran B, Clotet B, Blanco J (2010) Changes in T-cell subsets in HIV-HCV-coinfected patients during pegylated interferon-alpha2a plus ribavirin treatment. Antivir Ther 15:333–342
Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M (1994) Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921
Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, Sheehan KC, Schreiber RD, Oldstone MB (2015) Blockade of interferon beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 17:653–661
Nganou-Makamdop K, Billingsley JM, Yaffe Z, O’Connor G, Tharp GK, Ransier A, Laboune F, Matus-Nicodemos R, Lerner A, Gharu L, Robertson JM, Ford ML, Schlapschy M, Kuhn N, Lensch A, Lifson J, Nason M, Skerra A, Schreiber G, Bosinger SE, Douek DC (2018) Type I IFN signaling blockade by a PASylated antagonist during chronic SIV infection suppresses specific inflammatory pathways but does not alter T cell activation or virus replication. PLoS Pathog 14:e1007246
Palesch D, Bosinger SE, Mavigner M, Billingsley JM, Mattingly C, Carnathan D, Paiardini M, Chahroudi A, Vanderford T, Silvestri G (2018) Short-term pIFN-alpha2a treatment does not significantly reduce the viral reservoir of SIV-infected, ART-treated rhesus macaques. J Virol. https://doi.org/10.1128/jvi.00279-18
Pestka S, Krause CD, Walter MR (2004) Interferons, interferon-like cytokines, and their receptors. Immunol Rev 202:8–32
Rothenberger MK, Keele BF, Wietgrefe SW, Fletcher CV, Beilman GJ, Chipman JG, Khoruts A, Estes JD, Anderson J, Callisto SP, Schmidt TE, Thorkelson A, Reilly C, Perkey K, Reimann TG, Utay NS, Nganou Makamdop K, Stevenson M, Douek DC, Haase AT, Schacker TW (2015) Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc Natl Acad Sci USA 112:E1126–E1134
Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, Levin D, Wijeyesinghe S, Makamdop KN, del Prete GQ, Hill BJ, Timmer JK, Reiss E, Yarden G, Darko S, Contijoch E, Todd JP, Silvestri G, Nason M, Norgren RB Jr, Keele BF, Rao S, Langer JA, Lifson JD, Schreiber G, Douek DC (2014) Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 511:601–605
Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485
Sereti I, Krebs SJ, Phanuphak N, Fletcher JL, Slike B, Pinyakorn S, O’Connell RJ, Rupert A, Chomont N, Valcour V, Kim JH, Robb ML, Michael NL, Douek DC, Ananworanich J, Utay NS, Rv254/Search RS, teams Sp (2017) Persistent, Albeit reduced, chronic inflammation in persons starting antiretroviral therapy in acute HIV infection. Clin Infect Dis 64:124–131
Soper A, Kimura I, Nagaoka S, Konno Y, Yamamoto K, Koyanagi Y, Sato K (2017) Type I interferon responses by HIV-1 infection: association with disease progression and control. Front Immunol 8:1823
Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, Lebedeva M, DeCamp A, Li D, Grove D, Self SG, Borrow P (2009) Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol 83:3719–3733
Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H (2011) Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146:980–991
Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB (2013) Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–211
Teijaro JR, Studer S, Leaf N, Kiosses WB, Nguyen N, Matsuki K, Negishi H, Taniguchi T, Oldstone MB, Rosen H (2016) S1PR1-mediated IFNAR1 degradation modulates plasmacytoid dendritic cell interferon-alpha autoamplification. Proc Natl Acad Sci USA 113:1351–1356
Utay NS, Douek DC (2016) Interferons and HIV infection: the good, the bad, and the ugly. Pathog Immun 1:107–116
Veazey RS, Pilch-Cooper HA, Hope TJ, Alter G, Carias AM, Sips M, Wang X, Rodriguez B, Sieg SF, Reich A, Wilkinson P, Cameron MJ, Lederman MM (2016) Prevention of SHIV transmission by topical IFN-beta treatment. Mucosal Immunol 9:1528–1536
Walsh KB, Teijaro JR, Wilker PR, Jatzek A, Fremgen DM, Das SC, Watanabe T, Hatta M, Shinya K, Suresh M, Kawaoka Y, Rosen H, Oldstone MB (2011) Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci USA 108:12018–12023
Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG (2013) Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–207
Yan N, Chen ZJ (2012) Intrinsic antiviral immunity. Nat Immunol 13:214–222
Zhen A, Rezek V, Youn C, Lam B, Chang N, Rick J, Carrillo M, Martin H, Kasparian S, Syed P, Rice N, Brooks DG, Kitchen SG (2017) Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J Clin Investig 127:260–268
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Animal and Human Rights Statement
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nganou-Makamdop, K., Douek, D.C. Manipulating the Interferon Signaling Pathway: Implications for HIV Infection. Virol. Sin. 34, 192–196 (2019). https://doi.org/10.1007/s12250-019-00085-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12250-019-00085-5