
Abstract
The cGAS-MITA pathway of cytosolic DNA sensing plays essential roles in immune response against pathogens that contain DNA or with DNA production in their life cycles. The cGAS-MITA pathway also detects leaked or aberrant accumulated self DNA in the cytoplasm under certain pathological conditions, such as virus induced cell death, DNA damage, mitochondria damage, gene mutations, which results in autoimmune diseases. Therefore, the cGAS-MITA pathway must be tightly controlled to ensure proper immune response against pathogens and to avoid autoimmune diseases. The regulation of cGAS-MITA pathway at MITA-level have been extensively explored and reviewed elsewhere, here we provide a summary and perspective on recent advances in understanding of the cGAS regulation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The innate immune system is the first line of defense against incoming pathogens. Recognition of pathogen-associated molecular patterns (PAMPs) is essential for initiating innate immune response and subsequent adaptive immune response. Germ-line-encoded pattern recognition receptors (PRRs) detect PAMPs in extracellular, endosomal compartment and cytoplasm, leading to a series of signaling events which subsequently result in induction of type I interferons and proinflammatory cytokines. While many types of PRRs have been well demonstrated, such as TLRs (Toll-like receptors), CLRs (C-type lectin receptors), NLRs (NOD-like receptors) and RLRs (RIG-I-like receptors), the cytosolic DNA sensing pathway is gradually uncovered in recent years (Cai et al. 2014; Takeuchi and Akira 2010). There are several DNA sensors that have been identified to date, such as DAI, AIM2, IFI16, DDX41, RNA polymerase III and cGAS (Chiu et al. 2009; Hornung et al. 2009; Nie and Wang 2013; Orzalli and Knipe 2014; Parvatiyar et al. 2012; Takaoka et al. 2007; Unterholzner et al. 2010). Subsequent studies, especially gene knockout studies, have demonstrated that the cyclic GMP-AMP (cGAMP) synthase (cGAS) is a predominant and general sensor of cytosolic DNA no matter it is pathogenic or self DNA (Gao et al. 2015; Sun et al. 2013; Wu et al. 2013).
cGAS is expressed in a wide variety of tissues but prominently expressed in immune tissues. There are two adjacent IFN-sensitive response elements (ISREs) in the promoter of the cGAS gene, making it an ISG that can be induced by type I IFN-stimulation (Ma et al. 2015a). cGAS not only detects cytoplasmic DNA produced by a large variety of DNA-containing pathogens, such as DNA viruses and bacteria, but also detects DNA produced during reverse transcription by retrovirus (Gao et al. 2013; Hansen et al. 2014; Herzner et al. 2015; Mankan et al. 2014). More interestingly, cGAS also displays important functions in immune defense against some RNA viruses with no DNA intermediates in their life cycles, although the precise mechanism is still unclear (Aguirre and Fernandez-Sesma 2017; Aguirre et al. 2017; Ma and Damania 2016; Schoggins et al. 2014). In addition, cGAS is essential for cellular senescence and antitumor immunity by sensing accumulated cytoplasmic DNA from DNA damage or tumor DNA taken up by phagocytes such as dendritic cells, respectively (Lau et al. 2015; Mackenzie et al. 2017; Wang et al. 2017a; Yang et al. 2017).
The cGAS-MITA Signaling Pathway
Upon binding to DNA, cGAS dimerizes and forms a 2:2 complex with DNA, which induces a conformational change in the active site of cGAS. Activated cGAS catalyzes the synthesis of 2′3′-cGAMP from ATP and GTP (Kranzusch et al. 2014; Li et al. 2013; Sun et al. 2013). cGAMP then serves as a second messenger, which binds to the endoplasmic reticulum–resident adaptor protein MITA (also known as STING and ERIS (Wu et al. 2013; Zhang et al. 2013). Binding of cGAMP triggers conformational changes of the MITA homodimer and its subsequent translocation from the endoplasmic reticulum (ER), through the Golgi apparatus, to perinuclear microsomal compartments, accompanied with the recruitment and activation of kinases TBK1 and IKKα/β, which subsequently activate the transcription factors IRF3 and NF-κB respectively, ultimately leading to induction of type I interferons (IFNs) and proinflammatory cytokines (Cai et al. 2014; Ishikawa and Barber 2008; Saitoh et al. 2009; Zhong et al. 2008).
The cGAS-MITA pathway is critically essential in host immune defense against incoming pathogens, however, aberrant activation of the cGAS-MITA pathway can also lead to autoimmune and inflammatory disease, such as Aicardi Goutieres syndrome (AGS) and STING-associated vasculopathy with onset in infancy (SAVI) (Chen et al. 2016b; Gao et al. 2015; Liu et al. 2014; Pokatayev et al. 2016). Thus, the cGAS-MITA pathway must be tightly regulated to maintain immune homeostasis. Since the regulation of MITA is relatively clear and has been discussed extensively elsewhere (Chen et al. 2016b; Ma and Damania 2016; Ran et al. 2014), here we focus on the regulatory mechanisms of cGAS.
Regulation of cGAS by Post-translational Modifications
Post-translational modifications (PTMs) including phosphorylation, ubiquitination, and glutamylation have been reported to play important roles in the regulation of the cGAS-MITA pathway. For examples, glutamylation of cGAS impairs its ability to bind to DNA (Xia et al. 2016), whereas phosphorylation and K63- or K27-linked polyubiquitination of MITA is important for its activation and recruitment of TBK1 and IRF3 (Ran et al. 2014; Tsuchida et al. 2010; Wang et al. 2014; Zhang et al. 2012).
Regulation of cGAS by Phosphorylation
Akt, a critical protein kinase involved in cellular metabolism, cancer development and other cell process, was demonstrated to phosphorylate the S305 or S291 of human or mouse cGAS, respectively (Seo et al. 2015). These sites are in close proximity to the catalytic site of cGAS. Therefore, phosphorylation of cGAS on these sites inhibits the catalytic activity of cGAS, resulting in decreased cGAMP synthesis, which in turn impedes the induction of type I interferons upon DNA stimulation and HSV-1 infection. Akt-mediated the phosphorylation of cGAS acts as a brake to control antiviral response, whether there is a phosphatase to reverse the phosphorylation and turn on cGAS-mediated signaling is an interesting question to be explored.
Regulation of cGAS by Ubiquitination
Ubiquitination is a general post-translational modification and plays essential roles in the regulation of innate immune signaling cascades (Bhoj and Chen 2009). Although it is well established that MITA is extensively regulated by ubiquitination, the role of ubiquitination on cGAS regulation is just starting to be characterized. The E3 ligase RNF185, expression of which is upregulated in Systemic Lupus Erythematosus (SLE) patients, has been shown to be involved in the regulation of cGAS activity (Wang et al. 2017b). RNF185 potentiates the activity of cGAS upon DNA virus infection by catalyzing the formation of K27-linked ubiquitination chains on K137/384 sites of cGAS, resulting in enhanced production of type I interferons. Knockdown of RNF185 or mutation of cGAS ubiquitination sites resulted in decreased production of cGAMP. However, how K27-linked ubiquitination of cGAS modulates its enzymatic activity is still unclear. Another study, which discussed in detail in the following text, showed that K48-linked ubiquitination of cGAS leads to its degradation in an autophagosome-dependent manner, although the E3 ubiquitin ligase responsible for this process is unknown (Chen et al. 2016a).
Regulation of cGAS by SUMOylation
Similar with ubiquitination, SUMOylation is also a reversible post-translational modification and plays essential roles in modulating innate immune responses. Recently, two independent groups provide new insights into the roles of SUMOylation in cGAS regulation. Hu et al. (2016) showed that SUMOylation of cGAS promotes its stability. In detail, TRIM38, function as an E3 SUMO ligase, together with SENP2, a SUMO-specific protease, catalyzes dynamic SUMOylation of cGAS at K231 and K479. TRIM38 targets cGAS for SUMOylation in uninfected cells and during the early phase of viral infection, which prevents its K48-linked ubiquitination and degradation. In the late phase of infection, cGAS is deSUMOylated by SENP2 and subsequently degraded via proteasome. The SUMOylation- and ubiquitination-dependent dynamic regulation of cGAS stability ensures an efficient triggering of the cGAS-MITA pathway in the early phase of DNA virus infection, as well as its timely termination upon resolution of infection. Notably, in the same study, MITA is shown to be similarly regulated by SUMOylation.
In another study, Cui et al. (2017) showed that SENP7, also a SUMO-specific protease, positively regulates cGAS-mediated signaling. They found that cGAS is SUMOylated on K335, K372 and K382. Given that these SUMOylation sites are located in either DNA-binding surfaces or dimerization interface of cGAS, SUMOylation of cGAS at these sites impairs both its DNA-binding ability and its self-association. SENP7 interacts dynamically with cGAS upon HSV-1 infection and deSUMOylates cGAS, thus turns on the activation of cGAS. Notably, there are some discrepancies between these two studies, such as distinct SUMOylation patterns and different roles of SUMOylation in cGAS-mediated cytosolic DNA sensing pathway. Probably, these discrepancies are caused by different moiety of SUMO. Hu et al. focused on E3 ligase TRIM38-mediated SUMO-1 modification of cGAS, whereas Cui et al. mainly focused on SUMO2/3 modification of cGAS without specific E3 ligases. It is not surprising that different SUMO modifications have different effects on cGAS. Further studies are necessary to clarify these confusions.
Regulation of cGAS by Glutamylation
Glutamylation is a post-translational modification which is catalyzed by tubulin tyrosine ligase-like (TTLLs) glutamylases and reversed by enzymes of the cytosolic carboxypeptidase (CCPs) family (Rogowski et al. 2010). Recently, Xia et al. (2016) found that cGAS is tightly regulated by glutamylation. Mice deficient in either CCP5 or CCP6 are susceptible to the infection of DNA viruses, as a result of the failed induction of type I interferons. They found that polyglutamylation mediated by TTLL6 at glu272 dampens the DNA binding activity of cGAS, whereas monoglutamylation mediated by TTLL4 at glu302 blocks the enzymatic activity of cGAS. On the contrary, CCP6 and CCP5, the counterparts of TTLL6 and TTLL4, mediate the deglutamylation of cGAS at glu272 and glu302, respectively, thus release the inhibitory effects of glutamylation. Through dynamic regulation of the glutamylation state of cGAS, TTLL4, TTLL6, CCP5 and CCP6 work cooperatively to modulate cGAS-mediated immune responses. However, how these enzymes are triggered or regulated to control the glutamylation and deglutamylation of cGAS is unclear. Further studies are needed to resolve this question and provide more insights for the understanding of cGAS regulation.
Regulation of cGAS by Crosstalk with Other Pathway
Crosstalk with Autophagy
It has also been shown that Beclin-1, an autophagy protein, negatively regulates cGAS-mediated innate immune responses (Liang et al. 2014). On one hand, Beclin-1 directly interacts with the NTase domain of cGAS in a DNA binding dependent manner through its CCD domain, which impairs the enzymatic activity of cGAS, resulting in decreased cGAMP synthesis and subsequent impairment of type I interferon induction by DNA virus infection. On the other hand, knockdown of cGAS decreases the formation of autophagosome upon DNA stimulation, which suggests that cGAS may be involved in the formation of autophagosome. By this way, beclin-1 enhances autophagy-mediated degradation of cytosolic pathogen DNA to prevent excessive cGAS activation. These findings showed that autophagy adopts two strategies, elimination of pathogen DNA or inhibition of cGAS enzymatic activity, to negatively regulate DNA sensing pathway to avoid excessive activation of immune response.
Interestingly, a recent study by Chen et al. (2016a) provides another piece of evidence for negative regulation of DNA sensing pathway by autophagy. They found that E3 ligase TRIM14 could stabilize cGAS by inhibiting the autophagic degradation of cGAS. The K48-linked ubiquitination of cGAS at K414 is a signal for p62, a cargo receptor of selective autophagy. Without virus infection, cGAS undergoes K48-linked ubiquitination at K414, leading to p62-dependent selective autophagic degradation of cGAS. Upon DNA virus infection, TRIM14, as an interferon-stimulated gene, is induced and recruits the deubiquitinating enzyme USP14 to cleave K48-linked ubiquitin chains of cGAS, thus inhibiting p62-cGAS interaction as well as the degradation of cGAS. These studies reveal a positive feedback regulation of cGAS-mediated signaling by TRIM14 and provide insights into the crosstalk between autophagy and type I IFN signaling in innate immunity.
Crosstalk with Inflammasome
Viral infection triggers the production of type I interferons and activation of inflammasomes. There are many examples of crosstalks between type I interferons production signaling and inflammasome activation to balance these two processes (Guarda et al. 2011; Zhang et al. 2014). Wang et al. (2017c) found that cGAS is inhibited by caspases, essential components of inflammasome. Upon binding to DNA, AIM2 forms a complex with ASC and caspase-1, leading to the activation of caspase-1 (Fernandes-Alnemri et al. 2009; Hornung et al. 2009). Activated caspase-1 directly binds to and cleaves cGAS at D140/157, resulting in reduced cGAMP production and type I IFN induction. Importantly, caspase-1 only cuts cGAS upon DNA virus infection but not RNA virus infection. Consequently, AIM2-, ASC-, and caspase-1-deficient mice display enhanced resistance to DNA virus infection. In addition, Caspase-4, 5, and 11 can cut cGAS in conditions of non-canonical inflammasome activation (Wang et al. 2017c). Their findings reveal a role for inflammasome in the regulation of cGAS-mediated induction of type I IFNs and provided insights into the crosstalk between DNA virus induced innate immune responses and inflammasome activation.
Regulation of cGAS by Viral Proteins
Viruses have evolved elaborate mechanisms to antagonize the innate immune system. Plenty of studies have demonstrated that MITA, an essential adaptor downstream cGAS, is targeted by various viruses for immune evasion (Ding et al. 2013; Kalamvoki and Roizman 2014; Ma et al. 2015b; Wu et al. 2015). Recent studies supply several lines of evidence that cGAS is also antagonized by virus for immune evasion.
It is well established that Kaposi’s sarcoma-associated herpesvirus (KSHV) LANA localizes to the nucleus of infected cells. However, an isoform of KSHV LANA lacking the N terminal, generated by internal translation initiation, is localized to the cytoplasm. This cytoplasmic isoform of KSHV LANA was demonstrated to recruit and antagonize cGAS (Zhang et al. 2016). Overexpression of this isoform binds to cGAS and reduces the phosphorylation of TBK1 and IRF3, resulting in impaired production of type I IFNs and antiviral response. However, the precise mechanism has not been fully elucidated yet.
In addition to cytoplasmic isoform of LANA, ORF52, a tegument protein of KSHV, inhibits DNA sensing of cGAS (Wu et al. 2015). The enzymatic activity of cGAS is inhibited in the presence of ORF52 upon DNA stimulation or DNA virus infection. ORF52 can bind to both DNA and cGAS, and these abilities are important for cGAS inhibition. Intriguingly, its binding to cGAS is not dependent on its binding to DNA. ORF52 is a conserved protein in gammaherpes-viruses and its homologs in MHV68, RRV and EBV can also inhibit cGAS, suggesting that this is a general mechanism for immune evasion by gammaherpesviruses.
Dengue virus (DENV) is a single positive-stranded RNA virus of the family Flaviviridae, without DNA production in their life cycle. Nevertheless, it is well established that cGAS plays important roles in innate immune responses against DENV infection. It is proposed that cGAS detects mitochondrial DNA released during DENV infection (Aguirre et al. 2017; Ma and Damania 2016; West et al. 2015). A recent study showed that cGAS is degraded during DENV infection (Aguirre et al. 2017). DENV encodes a protease cofactor NS2B that promotes cGAS degradation in an autophagy–lysosome-dependent manner to avoid the detection of mitochondrial DNA, which results in the inhibition of type I interferon production during DENV infection. It is worthy to note that DENV can also evade the innate immune response through cleavage of MITA by its NS2B3 protease (Aguirre et al. 2012; Yu et al. 2012). Therefore, DENV targets both cGAS and MITA to minimize the host innate immune antiviral response.
Conclusions and Outlook
Since cGAS acts as an important cytosolic DNA sensor that leads to activation of innate immune antiviral responses, the activity of cGAS needs to be tightly modulated spatially and temporally by host factors to ensure the strength and duration of the immune responses. On the other hand, viruses have evolved elaborate mechanisms to antagonize the innate immune system. The cGAS-mediated signaling is targeted at every levels of signaling cascades by viruses. This review summarized the regulatory mechanisms of cGAS. The information presented in this review is summarized in Fig. 1 and Table 1. So far, our understanding of cGAS regulation is relatively limited. PQBP1 was reported to function as a coreceptor of cGAS in the sensing of retroviruses (Yoh et al. 2015), and studies also showed that IFI16 and cGAS work cooperatively during DNA virus infection in human foreskin fibroblasts (HFFs) and human keratinocytes (Almine et al. 2017; Orzalli et al. 2015). These studies indicate that the relationship between cGAS and other cytosolic DNA sensors needs to be further clarified, and also prompt us that there may be other coreceptor(s) or coactivator(s) of cGAS.

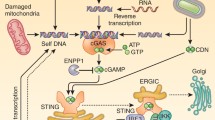
Schematic regulating network of cGAS. Detailed regulating proteins and virus antagonist of cGAS are depicted. Upon DNA binding to cGAS, cGAMP is synthesized by cGAS from ATP and GTP. Then binding of cGAMP to the adapter MITA induces conformational changes of MITA homodimer, leading to production of cytokines including type I interferons. Green arrows indicate positive regulators of the corresponding process. Red arrows indicate negative regulators of the corresponding process. Upon inflammasome activation, caspase-1 inhibits the cGAS-MITA pathway by cleavage of cGAS. DENV NS2B protein promotes the autophagy–lysosome-dependent degradation of cGAS. TRIM14 and USP14 inhibit the degradation of cGAS by cleaving K48-linked ubiquitin chains of cGAS. Beclin-1 promotes the autophagic degradation of cGAS. CCP5/6, TRIM38, SENP7 and RNF185 promote the activity of cGAS by deglutamylation, SUMOylation, deSUMOylation, ubiquitination of cGAS, respectively. TTLL4, TTLL6 and Akt inhibit the activation of cGAS by monoglutamylation, polyglutamylation, phosphorylation of cGAS, respectively. LANA of KSHV interacts and antagonizes cGAS. ORF52 of KSHV interacts with cGAS and inhibits the enzymatic activity of cGAS.
cGAS is tightly regulated by post-translational modifications, such as phosphorylation, ubiquitination, SUMOylation, and glutamylation. However, there still are some questions to be answered. Firstly, it is demonstrated that cGAS undergoes ubiquitin-dependent degradation, but the E3 ubiquitin ligases responsible for this process is still unknown. Secondly, the roles of dynamic SUMOylation of cGAS need further clarification. Thirdly, whether other PTMs, such as acetylation, glycosylation, which are also responsible for the regulation of cGAS are worthy to explore. Most importantly, how these PTMs work cooperatively to modulate the initiation, duration, and termination of cGAS-mediated signaling is pivotal. Systemic study on these dynamic processes of PTMs in vivo during virus infection will provide more exciting and convincing insights into cGAS-mediated signaling.
Finally, the roles of cGAS in autoimmune diseases and antitumor immunity have drawn more and more attention, but the regulation mechanisms of cGAS in these processes are still largely unknown. Further investigation of these questions will certainly shed new lights on our understanding of cGAS-mediated DNA sensing pathways and help to develop strategies to prevent and treat the related diseases.
Abbreviations
- PAMP:
-
Pathogen-associated molecular pattern
- PRR:
-
Pattern recognition receptor
- TLR:
-
Toll-like receptor
- CLR:
-
C-type lectin receptor
- RLR:
-
RIG-I-like receptor
- ER:
-
Endoplasmic reticulum
- TBK1:
-
TANK-binding kinase 1
- IKK:
-
IκB kinase
- IRF3:
-
Interferon regulatory factor 3
- HSV-1:
-
Herpes simplex virus 1
- cGAMP:
-
Cyclic GMP-AMP
- cGAS:
-
Cyclic GMP-AMP synthase
- IFN:
-
Interferon
- ISG:
-
Interferon-stimulated gene
- ISRE:
-
IFN-sensitive response element
- AGS:
-
Aicardi Goutieres syndrome
- SAVI:
-
STING-associated vasculopathy with onset in infancy
- PTM:
-
Post-translational modification
- SLE:
-
Systemic lupus erythematosus
- TTLL:
-
Tubulin tyrosine ligase-like
- CCP:
-
Cytosolic carboxypeptidase
- KSHV:
-
Kaposi’s sarcoma-associated herpesvirus
- MHV68:
-
Murine gammaherpesvirus-68
- RRV:
-
Ross River virus
- EBV:
-
Epstein–Barr virus
- HFF:
-
Human foreskin fibroblast
References
Aguirre S, Fernandez-Sesma A (2017) Collateral damage during dengue virus infection: making sense of DNA by cGAS. J Virol 91(4):e01081
Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, Rodriguez-Madoz JR, Mulder LC, Barber GN, Fernandez-Sesma A (2012) DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog 8:e1002934
Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, Fredericks AC, Tripathi S, Zhu T, Pintado-Silva J, Webb LG, Bernal-Rubio D, Solovyov A, Greenbaum B, Simon V, Basler CF, Mulder LC, Garcia-Sastre A, Fernandez-Sesma A (2017) Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol 2:17037
Almine JF, O’Hare CA, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, Beard PM, Unterholzner L (2017) IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 8:14392
Bhoj VG, Chen ZJ (2009) Ubiquitylation in innate and adaptive immunity. Nature 458:430–437
Cai X, Chiu YH, Chen ZJ (2014) The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54:289–296
Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, Zhou Y, Chen Y, Huang J, Wang RF, Cui J (2016) TRIM14 Inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol Cell 64:105–119
Chen Q, Sun L, Chen ZJ (2016) Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17:1142–1149
Chiu YH, Macmillan JB, Chen ZJ (2009) RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–591
Cui Y, Yu H, Zheng X, Peng R, Wang Q, Zhou Y, Wang R, Wang J, Qu B, Shen N, Guo Q, Liu X, Wang C (2017) SENP7 potentiates cGAS activation by relieving SUMO-mediated inhibition of cytosolic DNA sensing. PLoS Pathog 13:e1006156
Ding Q, Cao X, Lu J, Huang B, Liu YJ, Kato N, Shu HB, Zhong J (2013) Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol 59:52–58
Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513
Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ (2013) Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906
Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, Chen ZJ (2015) Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci USA 112:E5699–E5705
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–223
Hansen K, Prabakaran T, Laustsen A, Jorgensen SE, Rahbaek SH, Jensen SB, Nielsen R, Leber JH, Decker T, Horan KA, Jakobsen MR, Paludan SR (2014) Listeria monocytogenes induces IFNbeta expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J 33:1654–1666
Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kubler K, Wittmann S, Gramberg T, Andreeva L, Hopfner KP, Mertens C, Zillinger T, Jin T, Xiao TS, Bartok E, Coch C, Ackermann D, Hornung V, Ludwig J, Barchet W, Hartmann G, Schlee M (2015) Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol 16:1025–1033
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518
Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, Yin L, Shu HB (2016) Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity 45:555–569
Ishikawa H, Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678
Kalamvoki M, Roizman B (2014) HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc Natl Acad Sci USA 111:E611–E617
Kranzusch PJ, Lee AS, Wilson SC, Solovykh MS, Vance RE, Berger JM, Doudna JA (2014) Structure-guided reprogramming of human cGAS dinucleotide linkage specificity. Cell 158:1011–1021
Lau L, Gray EE, Brunette RL, Stetson DB (2015) DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350:568–571
Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, Li P (2013) Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39:1019–1031
Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, Shi M, Leslie BJ, Hopfner KP, Ha T, Oh BH, Jung JU (2014) Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 15:228–238
Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CC, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R (2014) Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371:507–518
Ma Z, Damania B (2016) The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe 19:150–158
Ma F, Li B, Liu SY, Iyer SS, Yu Y, Wu A, Cheng G (2015a) Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J Immunol 194:1545–1554
Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, Barber GN, Glaunsinger BA, Dittmer DP, Damania B (2015b) Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci USA 112:E4306–E4315
Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, Osborn RT, Wheeler AP, Nowotny M, Gilbert N, Chandra T, Reijns MAM, Jackson AP (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nat 548:461–465
Mankan AK, Schmidt T, Chauhan D, Goldeck M, Honing K, Gaidt M, Kubarenko AV, Andreeva L, Hopfner KP, Hornung V (2014) Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J 33:2937–2946
Nie Y, Wang YY (2013) Innate immune responses to DNA viruses. Protein Cell 4:1–7
Orzalli MH, Knipe DM (2014) Cellular sensing of viral DNA and viral evasion mechanisms. Annu Rev Microbiol 68:477–492
Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM (2015) cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci USA 112:E1773–E1781
Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS, Zaver SA, Schenk M, Zeng S, Zhong W, Liu ZJ, Modlin RL, Liu YJ, Cheng G (2012) The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol 13:1155–1161
Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, Morris HD, Yan N, Crouch RJ (2016) RNase H2 catalytic core Aicardi-Goutieres syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J Exp Med 213:329–336
Ran Y, Shu HB, Wang YY (2014) MITA/STING: a central and multifaceted mediator in innate immune response. Cytokine Growth Factor Rev 25:631–639
Rogowski K, van Dijk J, Magiera MM, Bosc C, Deloulme JC, Bosson A, Peris L, Gold ND, Lacroix B, Bosch Grau M, Bec N, Larroque C, Desagher S, Holzer M, Andrieux A, Moutin MJ, Janke C (2010) A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 143:564–578
Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S (2009) Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 106:20842–20846
Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM (2014) Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695
Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, Yuan W, Feng P, Park HS, Jung JU (2015) Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep 13:440–449
Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791
Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T (2007) DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S (2010) The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33:765–776
Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004
Wang Q, Liu X, Cui Y, Tang Y, Chen W, Li S, Yu H, Pan Y, Wang C (2014) The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41:919–933
Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ (2017a) cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA 114:1637–1642
Wang Q, Huang L, Hong Z, Lv Z, Mao Z, Tang Y, Kong X, Li S, Cui Y, Liu H, Zhang L, Zhang X, Jiang L, Wang C, Zhou Q (2017b) The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog 13:e1006264
Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, Zhou X, Gao J, Fang R, Meng G, Su X, Jiang Z (2017c) Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46:393–404
West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520:553–557
Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830
Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, Konrad A, Neipel F, Sturzl M, Whitby D, Li H, Zhu F (2015) Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18:333–344
Xia P, Ye B, Wang S, Zhu X, Du Y, Xiong Z, Tian Y, Fan Z (2016) Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat Immunol 17:369–378
Yang H, Wang H, Ren J, Chen Q, Chen ZJ (2017) cGAS is essential for cellular senescence. Proc Natl Acad Sci USA 114:E4612–E4620
Yoh SM, Schneider M, Seifried J, Soonthornvacharin S, Akleh RE, Olivieri KC, De Jesus PD, Ruan C, de Castro E, Ruiz PA, Germanaud D, des Portes V, Garcia-Sastre A, Konig R, Chanda SK (2015) PQBP1 is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell 161:1293–1305
Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, Lin YL (2012) Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog 8:e1002780
Zhang J, Hu MM, Wang YY, Shu HB (2012) TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J Biol Chem 287:28646–28655
Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ (2013) Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 51:226–235
Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, Zhang Z, Schneider M, Jiang Y, Fitzgerald KA, Ouyang S, Liu ZJ, Damania B, Shu HB, Duncan JA, Ting JP (2014) NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity 40:329–341
Zhang G, Chan B, Samarina N, Abere B, Weidner-Glunde M, Buch A, Pich A, Brinkmann MM, Schulz TF (2016) Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci USA 113:E1034–E1043
Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB (2008) The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550
Acknowledgements
Work in the authors’ laboratories is supported by the National Natural Science Foundation of China (31400742). The figure in this review was illustrated using the Servier Medical Art library with a slight modification under CC BY 3.0 license. http://smart.servier.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no conflict of interest.
Animal and Human Rights Statement
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Xiong, M., Wang, S., Wang, YY. et al. The Regulation of cGAS. Virol. Sin. 33, 117–124 (2018). https://doi.org/10.1007/s12250-018-0005-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12250-018-0005-6