
Abstract
Purpose
Gastroretentive drug delivery systems (GRDDS) have attracted interest for enhancement of absorption and bioavailability of some drugs. Itopride hydrochloride (ITOP) is a drug used for treatment of gastroesophageal reflux and other gastric motility disorders, but is characterized by narrow absorption window and short in vivo half-life. Therefore, it is expected that its formulation in expanding gastroretentive tablets would increase its gastric residence, thus leading to decreased frequency of administration and increased patient compliance.
Methods
The direct compression method was used for formulation of tablets. Four different hydrophilic polymers (xanthan gum, sodium alginate, gellan gum, pectin) were screened separately with Avicel 102 and PVP k30 as excipients. The effect of different factors (polymer type and amount, and excipient amount) on the tablet properties such as hardness, friability, thickness, diameter, weight variation, swelling, and in vitro drug dissolution was studied. In addition, swelling test, accelerated stability test, and in vivo study were performed on the optimized formulation.
Results
Tablets prepared using xanthan gum exhibited favorable properties compared to tablets prepared using the other gums, however increasing the polymer amount led to increased tablet friability. The selected formulation exhibited obvious expansion reaching 17.45 mm and lasting for 24 h, coupled with a sustained release behavior. X-ray scans in human volunteers suggested the residence of the tablet in the stomach for a period of 6 h in fed state.
Conclusion
Successful preparation of directly compressible ITOP expanding tablets was achieved in this study, which is expected to result in better therapeutic outcome in gastroesophageal reflux.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
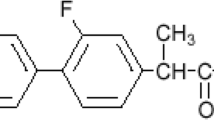
Gastroretentive drug delivery systems (GRDDS) are modified release drug delivery systems with increased gastric residence time in order to improve drug bioavailability [1,2,3,4,5]. Itopride hydrochloride (ITOP) is a prokinetic agent that activates gastric motility and improves functional dyspepsia by inhibiting acetyl cholinesterase enzyme and exhibiting an antagonist effect on dopamine D2 receptor. The therapeutic dose of ITOP is 50 mg to be taken three times daily, with elimination half-life 6 h and systemic availability 60% [6,7,8]. Owing to its high polarity, ITOP does not cause any CNS-related side effects due to its inability to cross the blood–brain barrier, making it superior to other similarly acting drugs [9, 10]. However, it suffers from narrow absorption window in the stomach and the upper part of the small intestine, leading to incomplete drug absorption and fast drug elimination [11,12,13,14]. The novelty of our work is the formulation of ITOP in expanding ablets, since to the best of our knowledge, no work has been attempted so far to formulate ITOP in this system. Formulating ITOP in would probably maximize its absorption, hence enhancing its therapeutic effectiveness and increasing patient compliance.
Several approaches have been employed to achieve gastric retention such as floating, swelling/expanding, bioadhesive, and other delayed gastric emptying systems. Several studies have developed different GRDDS of ITOP such as floating micro balloons [11, 15] and effervescent floating tablets [12, 13, 16].
Expanding systems represent an example of GRDDS, in which the expansion inside the stomach occurs either by unfolding or swelling. Unfolding is caused by mechanical shape memory, as the polymer and drug are in a folded state (e.g., inside the capsule), and upon contact with the gastric fluid, the capsule gelatin dissolves and releases the mechanically expanded configuration [17, 18]. For the swelling/expanding approach, a wide range of gums have been used as hydrophilic polymer matrices.
El-Zahaby et al. [19] formulated size expanding gastroretentive (GR) tablets of levofloxacin hemihydrate using low acyl gellan gum (LAGG), sodium alginate (SA), high methoxy pectin (HM pectin), and xanthan gum (XG). Jadhav et al. [20] formulated expanding GR tablets of diltiazem hydrochloride using HPMC and sodium CMC. The utilization of hydrophilic polymers in the expanding matrix allows the diffusion of gastrointestinal fluid through the drug formulation, resulting in swelling of the system to a size which exceeds the pyloric sphincter, hence causing retention of the expanded formulation inside the stomach for a prolonged time. This system is also termed a “plug type system” [21].
The matrix-forming hydrophilic polymers chosen for this work were XG, HM pectin, SA, and LAGG since they have been previously used as a GR matrix [19] and owing to their intricate molecular structure, high tensile strength, and high erosion-resistant matrix. The soluble salt form of these naturally charged polysaccharides causes higher swelling ability and lower erosion rate, in addition to their ability to ion-pair with oppositely charged drugs or excipients, forming a prolonged release matrix [22]. Regarding XG, it is soluble in water, and the viscosity of its solution is maintained over a wide range of pH and temperatures [23]. SA forms a gel through cross-linking or by pH adjustment [2]. LAGG forms a gel in presence of ions or high concentration of gums [24, 25]. Regarding pectin, its gelation depends on the type of pectin polymer, in which HM pectin needs a narrow range of pH and also the presence of a low amount of soluble solids, while low methoxy (LM) pectin needs divalent cations [26]. As previously reported, HM pectin can form a gel without the presence of cross-linker [27]. Therefore, the aim of this work is to formulate sustained release matrix of ITOP in an expanding/swelling system using the aforementioned polymers, to increase its residence time in the stomach and maximize its absorption.
Materials and Methods
Materials
ITOP was kindly supplied by Mash pharmaceutical Company, Egypt. HM Pectin (degree of esterification 55–65%) and LAGG were kindly supplied by CP Kelco, Denmark. SA (viscosity: 100–400 cps) was kindly supplied by Memphis pharmaceutical company, Egypt. XG (viscosity:1400–1600 mPas), Microcrystalline cellulose (Avicel 102), polyvinylpyrrolidone (PVP k30), and magnesium stearate were kindly supplied by EGY PHAR pharmaceutical company, Egypt. Hydrochloric acid (HCL) and barium sulfate were purchased from El Nasr Company, Egypt.
Methods
Preparation of Expanding ITOP Tablets
ITOP expanding tablets were prepared using the direct compression method [12, 16], which has the advantages of fewer processing steps and no involvement of moisture and heat [28]. Table 1 shows the composition of the different formulations, each formulation contained one type of hydrophilic polymer along with different concentrations of excipients (Avicel 102 as diluent, and PVP k30 as binder). Based on previous studies, the gums were used in concentrations of 45 or 72 w/w% of the total tablet weight [19]. Avicel 102 and PVP k30 were used in different proportions 100:0, 75:25, 50:50, 25:75, 0:100% w/w [29]. The components of each formulation were mixed by geometric dilution with mortar and pestle for 15 min till a homogenous mixture was obtained, and then the mixture was passed through sieve number 20, and finally magnesium stearate was added as a lubricant.
The powder mixture was manually fed and compressed using 12 mm flat face punch on single compression machine at 20 kN (model Karishma Pharma Machines 4040, Mumbai, India), based on a preliminary conducted study.
Pre-compression Assessment
Differential scanning calorimetry (DSC) method was used to detect any physicochemical interaction and incompatibility between ITOP and the utilized excipients. Samples were prepared in a drug: excipient ratio (1:1) [30, 31], in which 0.5 g of the drug and 0.5 g of individual excipients (either pectin, xanthan gum, sodium alginate, gellan gum, Avicel 102, and PVP were weighed, and each mixture was heated in a platinum pan at a rate of 10 °C/min to a temperature of 300 °C using indium as reference in a nitrogen atmosphere.
In addition, the flow properties of different formulation mixtures were evaluated for angle of repose, Carr’s index (C.I.%), and Hausner’s ratio (H.R.%) [13, 32,33,34].
Post-compression Parameters
Determination of Tablet Friability Percentage (%F), Hardness, Thickness, Diameter, and Weight Variation
For testing tablet friability percentage, twenty tablets were accurately weighed after de-dusting and placed in the drum of the friabilator (Copley, UK) at 25 rpm for 4 min, then the tablets were removed, brushed, and re-weighed for calculation of %F.
Ten tablets of each formulation were assessed for thickness and diameter using Vernier caliper and hardness using a hardness tester (Copley, UK). The weight variation test was performed by weighing ten tablets individually and calculating the average weight and standard deviation [12, 19].
In Vitro Drug Release Study
Selected tablet formulations were placed in 900 ml 0.1 N HCl as dissolution medium at 37 °C ± 0.5 and tested for their drug release profiles using a USP paddle apparatus type II (SR8 Plus, USA) at 50 rpm for 24 h. Aliquots of 5 ml were withdrawn at specified time intervals, filtered, and replenished with 5 ml fresh dissolution medium. The absorbance of each sample was measured at λ max 258 nm using a UV spectrophotometer after suitable dilution, and the cumulative % of ITOP released at each time interval was calculated [13, 35]. The kinetics of drug release from the different formulated tablets was determined through finding whether the release data fitted best to zero, first order, Korsmeyer-Peppas matrix or Higuchi [13, 15].
Swelling Study
Three tablets were initially weighed (W1), and then each tablet was placed in a beaker containing 50 ml of 0.1N HCl at 37 °C in a thermostatically controlled water bath [20, 30], and the weight was recorded at specified time intervals (0.25, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, and 6 h) or till the complete loss of tablet integrity before 6 h. The procedure was conducted by removing the tablets from the test medium and drying between two filter papers to remove excess surface water then reweighing (W2). The percent of water uptake was calculated using this equation
The tablets’ shape and integrity were observed during the experiment to investigate the morphological changes during tablet swelling. The diameters of the selected tablets were measured after swelling at time intervals: 6, 8, 12, and 24 h using a Vernier caliper, to confirm their ability to reside in the stomach after swelling.
Content Uniformity
The content uniformity of the selected tablet formulation was studied at the end of the compression step. Three tablets were crushed individually; each tablet was dissolved in 50 ml 0.1 N HCl and stirred for 10 min, which was sufficient to dissolve ITOP [12], since its solubility in pH 1.2 is 50 mg/ml. The solution was centrifuged, and an aliquot of supernatant was diluted and measured for estimating the drug content percentage [36].
Stability Study
The selected formulation was stored in a closed glass bottle in a stability cabinet at accelerated conditions (40 °C/75% RH) and evaluated after 3 months for drug assay and in vitro drug release [37]. The % drug release before and after storage were compared using four time points: an early one to exclude dose dumping and intermediate/final time points to assure complete release of the drug [38]. The difference factor (F1) was calculated to assess the percent (%) difference at each time point, and the relative error between the two profiles and the similarity factor (F2) were calculated to determine the % similarity between the two dissolution profiles [37, 39,40,41]. The calculated F1 and F2 values for the two profiles should be less than 15 and more than 50, respectively, for tablets having similar in vitro dissolution profiles.
In Vivo Study
The in vivo X-ray study was performed on two healthy male volunteers (of age 43 and 45 years) in both fasting and fed state at specific intervals 0.5, 1.5, 4, and 6 h. X-ray photographing technique (Genesis 50, Switzerland) on the abdominal region was used to observe the position and behavior of the selected expanding tablets and to verify their residence in the stomach at both erect and supine postures. The tablets used for the in vivo X-ray study were prepared by replacing the drug with the same amount of barium sulfate as a radio opaque material and attempting the same direct compression procedure [25, 42]. The volunteers were instructed not to take any medications 24 h before starting the experiment. For the fasting state, the volunteers fasted overnight (about 6 h), then ingested the tablet with 200 ml of water, and no food was allowed during the study time, with administration of 200 ml of water in need. For the fed state, high calorie–high fat breakfast composed of bread, milk, and eggs (900 cal) was given to the volunteers; then, after 2 h, the tablet was administered with 200 ml of water, and no food was allowed during the study time, with administration of 200 ml of water in need. The study was done under the supervision of a radiologist. PaxeraViewer software was used to analyze the X-ray images. The protocol of this study was approved by the research ethics committee of the faculty of pharmacy, Ain Shams University, approval number REC-ASU 48, following the ethical standards of the declaration of Helsinki for human experimentation.
Statistical Analysis
Results of the friability, hardness, thickness, diameter, weight variation, in vitro release, swelling, content uniformity, and stability were expressed as mean ± SD. All data were shown to pass the normality test. One-way analysis of variance (ANOVA) was used for comparison of data to determine their level of significance, followed by Tukey–Kramer post-test at level of significance p < 0.05 using GraphPad Instat program.
Results and Discussion
Pre-compression Assessment
A DSC study was performed on ITOP and the excipients to elucidate any physicochemical interaction that might affect the activity of drug. As shown in Fig. 1, the drug showed a significant sharp endothermic peak at 192.84 °C, corresponding to its reported melting temperature [11, 43]. No significant changes occurred to the melting peak of ITOP with pectin gum, LAGG, XG, and Avicel 102, while only a slight non-significant shift in the peak of ITOP occurred with PVP k30, and SA gum, which may be attributed to the mixing process, which lowers the concentration of each component in the mixture, thus resulting in slightly broader and lower melting points, but not truly representing any incompatibility [43].

DSC thermograms of a ITOP, b ITOP:HM pectin gum, c ITOP:LAGG, d ITOP:PVPK30, e ITOP:XG, f ITOP:SA, g ITOP:Avicel 102
Regarding the powder flow properties, all prepared powder mixtures exhibited good to excellent flow properties, with angle of repose values ranging from 21.8o to 28.39°, C.I.% ≤ 18, and H.R.% of < 1.25 except for pectin powders which exhibited only fair flowability, which may be attributed to the irregular shape of pectin particles resulting in low particles rearrangement and hence fair flowability [44, 45].
Post-compression Parameters
Determination of %F, Hardness, Thickness, Diameter, and Weight Variation of Tablets
As shown in Table 1, for the formulations G1-G10 prepared using LAGG, all tablets displayed %F less than 1% except for G1 and G2, which may be attributed to the absence of binder in G1 and its low percent in G2. Concerning formulations P1–P10 prepared using HM pectin, the majority of the formulations displayed %F > 1%, which may be attributed to the high percent of gum in (P1–P5) which consolidates by elastic deformation (temporary deformation) [44], and the inherent poor compactibility of pectin, with lack of plastic deformation in direct compression [46]. P6 formulation lacking PVP k30 gave a loosely bound tablet with high %F, and P7 formulation contained low percentage of binder, which was not sufficient for the formation of coherent tablets. Only formulations P8–P10 containing low percentage of pectin and high percentage of binder were found to be of good appearance and cohesiveness, with %F less than 1%. For SA gum containing formulations (A1–A10), the %F results were > 1% in the formulations containing no PVP k30 (A1, A6), or low amount of PVP k30 (A2), or no Avicel 102 (A10). The absence of binder or diluent resulted in high friability, which may be attributed to the high elastic recovery of alginate matrix associated with capping and lamination [47]. The remaining SA containing formulations showed %F < 1%. Similar to pectin containing tablets, XG containing formulations displayed %F > 1% in the formulations X1–X5, which could be attributed to the higher percentage of XG which exhibits low compactibility and brittle fracture behavior during compression [48], and the absence of PVP k30 in X6 may have hindered the formation of compact tablets [48, 49]. The low %F in formulations X7-X10 could be attributed to the lower percent of XG. The aforementioned results suggest that Avicel 102 and PVP k30 were important in enhancing the compression behavior of tablets and preserving their integrity, hence providing compact tablets with lower friability.
The hardness results of all prepared tablet formulations ranged from 2.56 to 24 Kp despite being compressed at the same force, which is attributed to the different percentage of polymers, binder, and diluent used [32]. All prepared tablets showed uniform thickness and diameter ranging from (3.90–4.26 mm) and (12.05–12.26 mm) respectively, and the weight variation for all the prepared tablets was found to be within the limit of 550 mg ± 5% (data not shown).
Based on the aforementioned data, the formulations with %F exceeding 1% were excluded from further characterization experiments.
In Vitro Drug Release Studies
Figure 2 shows the release profiles of ITOP from the different tablets’ formulations passing the friability test. From the data, it was found that the polymer type was the main factor affecting the drug release from the matrix, which was in the following order: LAGG > HM pectin > SA gum > XG. The fastest release obtained with LAGG tablets (100% release in less than 6 h) concurred with ElZahaby et al. [19] who reported similar release profile of tablets containing 60% w/w LAGG. Moreover, Elmowafy et al. [30] reported that a formulation containing 62.5%w/w of LAGG was completely released within 30 min. This was attributed to the ionization of the polysaccharide in 0.1N HCL which leads to electrostatic repulsion hindering gel formation along with tablet disintegration [50]. It was also reported that the high hydrophilicity of LAGG resulted in instant swelling, high disintegration tendency, and lower gelling ability [51, 52].

Release profiles of ITOP from different expanding tablets containing a LAGG, b HM pectin, c SA, d XG
All pectin formulations (P8–P10) exhibited early disintegration and could not maintain their physical integrity during the period of the release experiment, which may be attributed to tablet erosion and disintegration that occurs at pH 1.2 with low swelling ability of pectin in this acidic medium owing to the interconversion of carboxylate anion to the carboxyl group, hence converting it into pectinic acid [53,54,55,56].
The SA containing tablets showed sustained release behavior, approaching 100% release after 20 h, with no significant difference in the cumulative percent released of ITOP from the formulations (p > 0.05). The sustained release nature of the SA matrix was reported before in several studies, owing to its swellable nature and formation of a continuous gel layer [19, 57,58,59]. The release of ITOP from XG containing formulations was even more sustained than SA formulations, as shown by the significantly lower (p < 0.05) percent of ITOP release from the former at similar polymer concentration 45%w/w (SA formulations released ≈ 88% at 12 h and XG formulations released ≈ 51% at the same time interval). The drug release from XG matrices is believed to occur by a combination of two mechanisms: diffusion through the swollen matrix and disintegration from the eroded matrix layers [60,61,62,63,64]. Table 2 shows the kinetic data of ITOP release from different tablets matrices. From the data, it was observed that the release data for all tablet formulations except G4 fitted Korsmeyer-Peppas equation; implying diffusion-controlled release, in which the water is expected to diffuse into the tablet’s polymeric matrix, leading to its swelling and consequent diffusion of ITOP through the swollen gel layer [19, 25, 53, 57].
Worthy to note is that we have loaded 50 mg of ITOP in the tablets, similar to the commercially available tablets. As shown in Fig. 2, we have achieved almost 100% release from the tablets over duration of 24 h during their expected residency in the stomach, which is expected to provide clinically meaningful results. Interestingly, other authors who have formulated ITOP in microballons [11] and effervescent floating tablets [12, 16] have also achieved almost 100% release over a period of 24 h.
Based on the aforementioned results, the formulations prepared using SA (A3-A9) and xanthan gum (X7-X10) were selected for further characterization.
Swelling Study
As shown in Supplementary 1, 2, and Fig. 3, all formulations containing SA or XG displayed a swelling behavior (presented by % water uptake). SA-containing tablets were hydrated at low pH and formed a gel matrix due to intermolecular binding, which led to an increase in their weight upon contact with the medium [65]. Visual inspection showed that the morphology of the gel around the tablet matrix was neither viscous nor adhesive but tough and rubbery texture which is in agreement with Sriamornsak et al. [57]. The swelling of XG-containing tablets proceeded till 6 h, while in SA-containing tablets, the swelling took place till 3.5 h for tablets A4–A9 and till 4 h for tablet A3, after which they lost their integrity. SA-containing tablets were distorted upon swelling, forming a cracked laminated tablet due to the pressure build up in acidic media and rupture of surface which encouraged tablet erosion [26], as demonstrated in Fig. 3.

Swelling percentage of ITOP expanding tablets at pH 1.2 for a SA and b XG containing tablets, with representative morphology of tablets showing cracking in the former, and maintenance of integrity in the later
On the other hand, for XG-containing tablets, rapid swelling occurred which was attributed to the formation of a gel layer with no lag time, through hydration of XG particles, preventing the tablet disintegration [66,67,68], coinciding with the results of other authors [62, 66, 69]. The in vitro drug release was inversely correlated with the swelling behavior of the XG-containing formulations, which was also previously reported by other authors [30]. As shown in Fig. 3, XG-containing tablets maintained their shape and integrity. The water uptake study showed that XG formulations exhibited more swelling percentage than SA formulations at similar polymer concentration 45%w/w, as SA formulations reached ≈ 116%w/w at 3.5 h and, on the other hand, XG formulations ≈ 245%w/w at the same time interval. Therefore, the SA-containing tablets were excluded from further characterization.
Based on the previous results, formulation X7 containing XG (45%w/w) in addition to Avicel 102: PVPK30 3:1 was selected for further analysis, since it exhibited sustainment of drug release accounting for 58.53% at 12 h and 94.37% in 24 h and maintained the physical integrity of the matrix throughout the whole 24 h duration, which met our target compared to its counterparts. Using the same procedure and conditions of the swelling test, X7 tablet diameter was measured initially and at 6, 8, 12, and 24 h after immersion in 0.1N HCL, and they were found to increase progressively from 12.01 ± 0.15 mm at 0 h to 16.66 ± 0.33, 17.08 ± 0.46, 17.57 ± 0.54, and 17.45 ± 0.54 mm at 6, 8, 12, and 24 h respectively, while still maintaining their integrity. This was advantageous in our case, since it was reported by other authors that a diameter of about 12–18 mm could achieve gastric retention for several hours by hindrance of the passage through the pyloric sphincter and allowing the retention of the drug formulation inside the stomach [17, 70].
Stability Study and Content Uniformity
To indicate the stability of the selected formulation (X7) under accelerated storage conditions, drug assay and in vitro release tests were performed after storage for 3 months at 40 °C/ 75% RH. The assay results before (T0) and after stability were 100.06 ± 2.39 and 98.82 ± 1.17%, respectively, which proved the stability of formulation, shown by the insignificant change in drug content (p > 0.05), and which also complied with USP 42, in which the drug content should be within 90–110% of the label claim.
For the in vitro release test, four selected points in the release profile were selected and compared with the original profile (USP42), as shown in Fig. 4. The difference factor F1 was calculated to be 4.58%, indicating no significant difference between the compared profiles, and the similarity factor F2 was calculated to be 68.28%, indicating high similarity between the two profiles, which further indicates the stability of the formulation.

Comparative release profiles of formulation X7 before and after accelerated stability storage
In Vivo Study
The X-ray radiographic technique has been used in many GRDDS studies to determine the gastric residence time of tablets despite its inability to give detailed images of the body, owing to its ability to track the position of tablets with time [42, 71, 72]. The study was carried out on volunteers in fed and fasting state, and from the X-ray radiographic images in Fig. 5, it is demonstrated that the residence time varied from the fasting to the fed state. In the fasting state, the selected tablet formulation X7 was observed in the stomach at 0.5 h and appeared in the small intestine after 1.5 h and then disappeared at 4 h. The rapid emptying of the tablet from the stomach at the fasted state may be attributed to the sweeping wave evacuating the upper GIT through migrating motor complex (MMC) pattern occurring every 1.5–2 h [42, 73]. On the other hand during the fed state, the selected tablet formulation X7 was observed in stomach at 0.5, 1.5, 4, and 6 h, which could be attributed to the slower gastric motility in fed state comparing to fasting state, as the presence of food delayed the evacuation of the stomach hence enhanced the gastric residence of selected formulation [74,75,76]. The X-ray photographs demonstrated that the tablets increased in size after 4 h, and at 6 h, the tablets appeared to be in the expanded form. These results were in agreement with Meka et al. [42] who reported that the tablets appeared in the small intestine after 3 h and disappeared at 6 h in the fasted state, while they remained in the stomach for 6 h in the fed state. This expansion caused by the swelling of the matrix in contact with the gastric fluid is assumed to help in the plugging of the pyloric sphincter and in preventing the tablets’ passage to the small intestine, hence leading to the increased retention time of tablets in the stomach [70]. It is worthy to note that regarding gastric emptying rate, it was proven to depend on the dosage form itself and the state of the stomach (fed or fasted state). The reported gastric emptying time ranges from 2 min to 2 h. The GIT motility involves two modes: the first one is called inter-digestive motility which takes place during the fasting state to evacuate the upper GIT, and the second one is the digestive motility which occurs during the fed state. The GIT motility during the fasting state is controlled by a pattern called migrating motor complex (MMC), which is a pattern of movement and silence controlled by motilin hormone and subdivided into four phases. The presence of food stops the MMC cycle and starts the digestive phase, and the food intake with an oral dosage form is known to increase its gastric retention. However, a factor of paramount importance is the size of the dosage form, in which the gastric residence time is directly proportional to the size of the dosage form. As the size increases the gastric residence time increases, since the relatively large size prevents the dosage form from passing through the pyloric sphincter. However, as observed from the results, despite the swellability of the tablets, the gastric retention was only physiological and not attributed to the swelling of the tablets, as it was not achieved in the fasting state.

X-ray photographs of gastric expanding tablets X7 in a representative volunteer. a Fasting state (I) 0.5 h (II) 1.5 h (III) 4 h. b Fed state (I) 0.5 h (II) 1.5 h (III) 4 h (IV) 6 h
While the results of this study suggest that expanding tablets could be promising delivery system for itopride, further optimization studies are still needed, especially in terms of establishing tablet compression profile, testing the swelling ability of the selected formulation across a pH range, and to further prepare tablets from xanthan gum of different molecular weights and viscosity values. Moreover, the inclusion of experimental controls in the in vivo study such as administration of tablets prepared without xanthan gum could have highlighted the gastric retention ability of the formulation. Moreover, since the in vivo study was only conducted on two volunteers for the proof of concept, a more extensive clinical study will be attempted in the future, to assess the retention as well as the pharmacokinetic parameters of the selected itopride tablet formulation and its therapeutic efficacy in treatment of gastroesophageal reflux.
Conclusion
An expanding matrix tablet of itopride was successfully prepared using the direct compression technique using hydrophilic matrix composed of xanthan gum, displaying high swelling ability in vitro, with high storage stability, and can be a promising dosage form for drugs with narrow absorption window.
Data Availability
All data supporting the findings of this study are available within the paper and its Supplementary Information.
References
Ibrahim M, Naguib YW, Sarhan HA, et al. Gastro-retentive oral drug delivery systems: a promising approach for narrow absorption window drugs. J Adv Biomedical Pharm Sci. 2019;2(3):98–110. https://doi.org/10.21608/JABPS.2019.11357.1042.
Patil S, Rathi M, Misra A. Applications of polymers in gastric drug delivery. In: Applications of Polymers in Drug Delivery (Second Edition). El-Sevier 2021. pp 77–104.
Vrettos NN, Roberts CJ, Zhu Z. Gastroretentive technologies in tandem with controlled-release strategies: a potent answer to oral drug bioavailability and patient compliance implications. Pharmaceutics. 2021;13(10):1591. https://doi.org/10.3390/pharmaceutics13101591.
Chaudhary S, Dua JS, Prasad DN. recent development in floating drug delivery system: an overview. J Drug Deliv Ther. 2022;12(1):185–93. https://doi.org/10.22270/jddt.v12i1.5171.
Jeganath DS. Recent approaches of gastroretentive drug delivery system – a review. Asian J Pharm. 2022;16(1):10–4. https://doi.org/10.22377/ajp.v16i1.4273.
Ganji A, Rao V, Mahalakshmi K, et al. Formulation and evaluation of sustained release tablets of itopride hydrochloride. Int Res J Pharm. 2013;4:70–4. https://doi.org/10.7897/2230-8407.041016.
Patel R, Patel K, Patel Z, et al. Formulation and evaluation of sustained release microspheres of itopride hydrochloride. AEGAEUM J. 2020;8:1913–23.
Liang L, Yu J, Xiao L, et al. Comparative efficacy of various pharmacological interventions in the treatment of functional dyspepsia: a network meta-analysis. Dig Dis Sci. 2021; Epub ahead of print. https://doi.org/10.1007/s10620-021-06846-1.
Huang X, Lv B, Zhang S, et al. Itopride therapy for functional dyspepsia: a meta-analysis. World J Gastroenterol. 2012;18(48):7371–7. https://doi.org/10.3748/wjg.v18.i48.7371.
Vandenberghe A, Schol J, Van den Houte K, et al. Current and emerging therapeutic options for the management of functional dyspepsia. Expert Opin Pharmacother. 2020;21(3):365–76. https://doi.org/10.1080/14656566.2019.1707805.
Bansal S, Beg S, Asthana A, et al. QbD-enabled systematic development of gastroretentive multiple-unit microballoons of itopride hydrochloride. Drug Deliv. 2014;23(2):437–51. https://doi.org/10.3109/10717544.2014.916771.
Ahmed SM, Ali AA, Ali AM, Hassan OA. Design and in vitro/in vivo evaluation of sustained-release floating tablets of itopride hydrochloride. Drug Design Devel Ther. 2016;10:4061. https://doi.org/10.2147/DDDT.S115909.
Mamatha A, Sharanya B. Formulation and in vitro evaluation of gastro retentive drug delivery system of prokinetic agent (itopride hydrochloride). IJPBS. 2018;8(2):491–503.
Rashmitha V, Pavani S, Rajani T, et al. An update on floating drug delivery system: a review. Int J Adv Pharm Biotechnol. 2020;6:9–18. https://doi.org/10.38111/ijapb.20200604003.
Hajare PP, Rachh PR. Gastroretentive microballoons: a novel approach for drug delivery. Int J Pharm Sci Res. 2020;11:1075–83. https://doi.org/10.13040/IJPSR.0975-8232.11(3).1075-83.
Chandira RM, Bhowmik D, Bhattacharjee C, et al. Formulation and evaluation of gastroretentive drug delivery system of gastroprokinetic drug itopride hydrochloride. Int J Pharm Pharm Sci. 2010;2:53–65.
Klausner EA, Lavy E, Friedman M, et al. Expandable gastroretentive dosage forms. J Controlled Rel. 2003;90(2):143–62. https://doi.org/10.1016/S0168-3659(03)00203-7.
Bhise MR, Sudke SG, et al. An extensive insight on gastroretentive drug delivery systems. PAIDEUMA J. 2020;13:59–71.
El-Zahaby SA, Kassem AA, El-Kamel AH, et al. Formulation and in vitro evaluation of size expanding gastro-retentive systems of levofloxacin hemihydrate. Int J Pharm. 2014;464(1–2):10–8. https://doi.org/10.1016/j.ijpharm.2014.01.024.
Jadhav RP, Kumbhar SB, Kengar MD, et al. Design and in-vitro evaluation expandable gastro retentive tablets of diltiazem hydrochloride. AJPTech. 2020;10(2):53–9. https://doi.org/10.5958/2231-5713.2020.00011.2.
Tripathi J, Thapa P, Maharjan R, et al. Current state and future perspectives on gastroretentive drug delivery systems. Pharmaceutics. 2019;11(4):193. https://doi.org/10.3390/pharmaceutics11040193.
Deshmukh AS, Aminabhavi TM. Pharmaceutical applications of various natural gums. In: Polysaccharides. Switzerland: Bioactivity and Biotechnology, 2015. pp 1933–1967.
Hasnain MS, Nayak AK. Xanthan gum in drug delivery applications. In: Natural Polysaccharides in Drug Delivery and Biomedical Applications: United kingdom (UK): Academic Press; 2019. pp 121–144.
Osmałek T, Froelich A, Tasarek S. Application of gellan gum in pharmacy and medicine. Int J Pharm. 2014;466(1–2):328–40. https://doi.org/10.1016/j.ijpharm.2014.03.038.
Karemore MN, Bali NR. Gellan gum based gastroretentive tablets for bioavailability enhancement of cilnidipine in human volunteers. Int J Biol Macromol. 2021;174:424–39. https://doi.org/10.1016/j.ijbiomac.2021.01.199.
Layek B, Mandal S. Natural polysaccharides for controlled delivery of oral therapeutics: a recent update. Carbohydr Polym. 2020;230:115617. https://doi.org/10.1016/j.carbpol.2019.115617.
Salbu L, Bauer-Brandl A, Tho IJAP. Direct compression behavior of low-and high- methoxylated pectins. AAPS PharmSciTech. 2010;11(1):18–26. https://doi.org/10.1208/s12249-009-9349-4.
Agiba AM, Abdel-Hamid S, Nasr M, et al. Geriatric-oriented high dose nutraceutical ODTs: formulation and physicomechanical characterization. Curr Drug Deliv. 2018;15(2):267–77. https://doi.org/10.2174/1567201814666170320143824.
Kusuma AP, Fudholi A, Nugroho AK, et al. Optimization direct compression’s co-processed excipient microcrystalline cellulose PH 102 and Povidone® K 30. IOSR J Pharm Biol Sci. 2014;9:65–9. https://doi.org/10.9790/3008-09246569.
Elmowafy EM, Awad GA, Mansour S, et al. Release mechanisms behind polysaccharides-based famotidine-controlled release matrix tablets. AAPS PharmSciTech. 2008;9(4):1230–9. https://doi.org/10.1208/s12249-008-9155-4.
Abouelatta SM, Aboelwafa AA, El-Gazayerly ON. Gastroretentive raft liquid delivery system as a new approach to release extension for carrier-mediated drug. Drug Deliv. 2018;25(1):1161–74. https://doi.org/10.1080/10717544.2018.1474969.
Khaled A, Abdel-Hamid S, Nasr M, et al. Fabrication of extended-dissolution divalproex tablets: a green solvent-free granulation technique. Drug Dev Ind Pharm. 2020;46(6):975–87. https://doi.org/10.1080/03639045.2020.1764023.
Bhosale AR, Shinde JV, Chavan RS, et al. A comprehensive review on floating drug delivery system (FDDS). J Drug Deliv Ther. 2020;10(6):174–82. https://doi.org/10.22270/jddt.v10i6.4461.
Setia M, Kumar K, Teotia D, et al. Gastro-retentive floating beads a new trend of drug delivery system. J Drug Deliv Ther. 2018;8(3):169–80. https://doi.org/10.22270/jddt.v8i3.1717.
Ratnaparkhi MP. Formulation and development of floating drug delivery of Itopride Hcl. J Drug Deliv Ther. 2013;3(4):222–8. https://doi.org/10.22270/jddt.v3i4.579.
Ali A, Iqbal M, Akhtar N, et al. Assessment of xanthan gum based sustained release matrix tablets containing highly water-soluble propranolol HCl. Acta Pol Pharm. 2013;70(2):283–9.
ICH Guidelines Q1A(R2). Stability testing of new drug substances and products; 2003. London: EMEA.
United States Pharmacopoeia 42 /The National Formulary 37. Rockville, MD: USP Convention; 2019.
Kassaye L, Genete G. Evaluation and comparison of in-vitro dissolution profiles for different brands of amoxicillin capsules. Afr Health Sci. 2013;13:369–75. https://doi.org/10.4314/ahs.v13i2.25.
Chawra HS, Tanwar Y, Singh S. Formulation and characterization of floating matrix tablets for an antihypertensive drug: valsartan. Pharma Innov J. 2018;7(9):9–16.
Gunda RK, Vijayalakshmi A. Development and evaulation of gastroretentive formulations for moxifloxacin hydrochloride. Thai J Pharm Sci. 2020;44(1):30–9.
Meka VS, Nali SR, Songa AS, et al. Statistical optimization of a novel excipient (CMEC) based gastro retentive floating tablets of propranolol HCl and its in vivo buoyancy characterization in healthy human volunteers. DARU J Pharm Sci. 2012;20(1):1–12. https://doi.org/10.1186/2008-2231-20-21.
Shah S, Madan S, Agrawal S. Formulation and evaluation of microsphere based oro dispersible tablets of itopride HCL. DARU J Pharm Sci. 2012;20(1):1–12. https://doi.org/10.1186/2008-2231-20-24.
Salbu L, Bauer-Brandl A, Alderborn G, Tho I. Effect of degree of methoxylation an4d particle size on compression properties and compactibility of pectin powders. Pharm Dev Technol. 2012;17(3):333–43. https://doi.org/10.3109/10837450.2010.535831.
Chomto P, Nunthanid J. Physicochemical and powder characteristics of various citrus pectins and their application for oral pharmaceutical tablets. Carbohydr Polym. 2017;174:25–31. https://doi.org/10.1016/j.carbpol.2017.06.049.
Assifaoui A, Chambin O. Pectin as drug-release vehicle. In: pectin: technological and physiological properties. Switzerland: Springer; 2020. pp 189–207.
Benabbas R, Sanchez-Ballester NM, Bataille B, et al. Structure-properties relationship in the evaluation of alginic acid functionality for tableting. AAPS PharmSciTech. 2020;21(3):1–11. https://doi.org/10.1208/s12249-020-1633-3.
Abu Fara D, Dadou SM, Rashid I, et al. A direct compression matrix made from xanthan gum and low molecular weight chitosan designed to improve compressibility in controlled release tablets. Pharmaceutics 2019;11(11)603. https://doi.org/10.3390/pharmaceutics11110603.
Luo Y, Hong Y, Shen L, et al. Multifunctional role of polyvinylpyrrolidone in pharmaceutical formulations. AAPS PharmSciTech. 2021;22(1):1–16. https://doi.org/10.1208/s12249-020-01909-4.
Norton A, Cox P, Spyropoulos F. Acid gelation of low acyl gellan gum relevant to self-structuring in the human stomach. Food Hydrocoll. 2011;25:1105–11. https://doi.org/10.1016/j.foodhyd.2010.10.007.
Gopinath H, Shanmugasundrama S, Bada PK. Disintegrants- brief review. J Chem Pharm Sci. 2012;5:105–22.
Bhatti S, Kaushik M. Utilization of natural super disintegrant in mouth dissolving tablet: a simplified review. IPP. 2020;8:32–8. https://doi.org/10.31690/ipp.2020.v08i02.004.
Sriamornsak P, Thirawong N, Weerapol Y, et al. Swelling and erosion of pectin matrix tablets and their impact on drug release behavior. Eur J Pharm Biopharm. 2007;67:211–9. https://doi.org/10.1016/j.ejpb.2006.12.014.
Akhgari A, Abbaspour M, Rezapour S, et al. Evaluation of swelling, erosion and drug release from polysaccharide matrix tablets based on pectin and inulin. Jundishapur J Nat Pharm Prod. 2011;6:51–8.
Sundar Raj A, Rubila S, Jayabalan R, et al. A review on pectin: chemistry due to general properties of pectin and its pharmaceutical uses. Sci Rep. 2012;1:550–3. https://doi.org/10.4172/scientificreports.550.
Gawkowska D, Cybulska J, Zdunek AJP. Structure-related gelling of pectins and linking with other natural compounds: a review. Polymers. 2018;10(7):762. https://doi.org/10.3390/polym10070762.
Sriamornsak P, Thirawong N, Korkerd K. Swelling, erosion and release behavior of alginate-based matrix tablets. Eur J Pharm Biopharm. 2007;66:435–50. https://doi.org/10.1016/j.ejpb.2006.12.003.
Saha T, Masum ZU, Ashrafi S. Preparation and in-vitro evaluation of sodium alginate based gastroretentive floating tablet of domperidone. Galore Int J Health Sci Res. 2018;3:1–4.
Davidovich-Pinhas M, Bianco-Peled H. A quantitative analysis of alginate swelling. Carbohydr Polym. 2010;79(4):1020–7. https://doi.org/10.1016/j.carbpol.2009.10.036.
Benny IS, Varadarajan G, Ponnusami V. Review on application of xanthan gum in drug delivery. Int J PharmTech Res. 2014;6:1322–6.
Zaman M, Akhtar F, Baseer A, et al. Formulation development and in-vitro evaluation of gastroretentive drug delivery system of loxoprofen sodium: a natural excipients-based approach. Pak J Pharm Sci. 2021;34:57–63. https://doi.org/10.36721/PJPS.2021.34.1.REG.057-063.1.
Pathan D, Memon S, Sayyed R. Formulation development and evaluation of gastroretentive floating drug delivery system using natural polymer. JCPR. 2018;8(3):2426–36.
Chinthaginjala H, Gandla CB, Challa MR, et al. Formulation and in vitro evaluation of floating tablets of dicloxacillin sodium using different polymers. J Young Pharm. 2019;11(3):247. https://doi.org/10.5530/jyp.2019.11.51.
Bose PS, Reddy PS, Ravi V, et al. Formulation and evaluation of sustained release floating tablets of diltiazem HCL using xanthan gum. Res J Pharm Biol Chem Sci. 2011;2:319–28.
Jain D, Bar-Shalom D. Alginate drug delivery systems: application in context of pharmaceutical and biomedical research. Drug Dev Ind Pharm. 2014;40(12):1576–84. https://doi.org/10.3109/03639045.2014.917657.
Talukdar MM, Kinget R. Swelling and drug release behaviour of xanthan gum matrix tablets. Int J Pharm. 1995;120:63–72. https://doi.org/10.1016/0378-5173(94)00410-7.
Badhan AC, Mashru RC, Shah PP, et al. Development and evaluation of sustained release gastroretentive minimatrices for effective treatment of H. pylori infection. AAPS PharmSciTech. 2009;10(2):459–67. https://doi.org/10.1208/s12249-009-9231-4.
Javaid MU, Zaman M, Batool D, et al. Formulation and in vitro analysis of gastro retentive drug delivery system containing timolol maleate. Acta Pol Pharm. 2017;74:1177–86.
Bhatta R, Hossain MS, Banik S, et al. Swelling and mucoadhesive behavior with drug release characteristics of gastoretentive drug delivery system based on a combination of natural gum and semi-synthetic polymers. Marmara Pharm J. 2018;22(2):286–98. https://doi.org/10.12991/mpj.2018.66.
Mahor A, Gupta R. Recent advances in gastro retentive drug delivery systems and its application on treatment of H. Pylori infections. J Anal Pharm Res. 2018;7(4):404–10. https://doi.org/10.15406/japlr.2018.07.00258.
Teaima M, Hamid MMA, Shoman NA, et al. Promising swellable floating bupropion tablets: formulation, in vitro/in vivo evaluation and comparative pharmacokinetic study in human volunteers. Drug Des Devel Ther. 2020;14:2741. https://doi.org/10.2147/DDDT.S258571.
Bomma R, Naidu RS, Yamsani M, et al. Development and evaluation of gastroretentive norfloxacin floating tablets. Acta Pharm. 2009;59(2):211–21. https://doi.org/10.2478/v10007-009-0019-6.
Singh R, Kakar S. Gastroretentive drug delivery systems: a review. African J Pharm Pharmacol. 2015;9(12):405–17. https://doi.org/10.5897/AJPP2015.4307.
Yusif R, Abu Hashim I, Mohamed E, et al. Investigation and evaluation of an in situ interpolymer complex of carbopol with polyvinylpyrrolidone as a matrix for gastroretentive tablets of ranitidine hydrochloride. Chem Pharm Bull. 2016;64:42–51. https://doi.org/10.1248/cpb.c15-00620.
Sapavatu SN, Jadi RK. Formulation development and characterization of gastroretentive drug delivery systems of loratadine. Int J Appl Pharm. 2019;11:91–9. https://doi.org/10.22159/ijap.2019v11i6.35194.
Jagdale SC, Agavekar AJ, Pandya SV, et al. Formulation and evaluation of gastroretentive drug delivery system of propranolol hydrochloride. AAPS PharmSciTech. 2009;10(3):1071–9. https://doi.org/10.1208/s12249-009-9300-8.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmed, W., El-Gogary, R.I., Nasr, M. et al. Development and in Vitro/In Vivo Evaluation of Itopride Hydrochloride Expanding Tablets. J Pharm Innov 18, 1350–1361 (2023). https://doi.org/10.1007/s12247-023-09729-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12247-023-09729-2