
Abstract
Purpose
The classic liquisolid technique is used to enhance the dissolution rate of poorly water-soluble drugs, but in some cases, it is impossible to reach the desired dissolution rate using this technique alone. Therefore, a novel approach using a combination of ball milling and liquisolid technology was investigated to improve the dissolution rate for celecoxib.
Methods
First, celecoxib was dispersed in a liquid vehicle (PEG 200), then ground in a ball mill for 3 h. Other excipients, including PVP, microcrystalline cellulose as the carrier powder, and silica as the coating material, were added to the mortar. Dissolution testing was carried out in simulated intestinal fluid (SIF) and simulated gastric fluid (SGF) media. The effects of aging on the hardness and dissolution profile were also studied. X-ray diffraction (XRD) and differential scanning calorimetry (DSC) was used to identify changes in the crystallinity or complex formation.
Results
The novel formulations showed a higher dissolution rate than the conventional tablet or classic liquisolid formulations. Aging did not affect the hardness and dissolution profiles of the liquisolid compacts. The DSC and XRD results suggested that the enhanced dissolution rate is not caused by the formation of any complexes and reduction in crystallinity degree may contribute to the dissolution enhancement. The enhanced dissolution rate is attributed to the elevated specific surface area of the drug in the liquisolid state.
Conclusion
The results showed that liquisolid technology combined with ball milling is an efficient tool for enhancing the dissolution of poorly water-soluble drugs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the pharmaceutical industry, each drug and its pharmaceutical dosage forms are developed and supplied to create a specific pharmacological effect. Oral solid pharmaceutical dosage forms need to initially dissolve in gastrointestinal fluid as part of the absorption process, which transports the drug into the circulatory system to produce a pharmacological effect. In the case of water-insoluble drugs that are administered orally, the rate of drug uptake is usually controlled by the rate of drug dissolution in gastrointestinal fluid, and in such cases, the rate of uptake will be equal to the rate of dissolution [1, 2]. Therefore, by increasing the rate of drug dissolution, the rate, and sometimes the extent, of drug absorption can be increased. The best and most practical way to improve the bioavailability of water-insoluble drugs is to increase the solubility and dissolution rate of the drug [3].
Celecoxib is a water-insoluble drug whose oral absorption is affected by its rate of dissolution. More specifically, the bioavailability of the drug is controlled by its dissolution rate in the gastrointestinal tract (GIT) [4]. Therefore, its bioavailability can be increased by increasing the drug dissolution rate in the GIT. The liquisolid technique is used to increase the dissolution rate of water-soluble drugs [5]. In liquisolid technique, a non-volatile solvent dissolves the poorly water-soluble drug and the resultant solution or suspension is known as liquid medication [6, 7]. The liquid medication (solution or suspension) is then converted to dry-looking, non-sticky, free-flowing powder by adding carrier and coating materials to make it a compactible powder mixture [6, 8, 9]. Liquisolid technique is one of the promising methods for dissolution enhancement of poorly water soluble drugs [10, 11] but it seems its application in high dose drugs is challenging [12].
The grinding method is another method to enhance the dissolution rate by reducing the particle size of the poorly water-soluble drugs, particularly when the poorly water-soluble drug is mixed with hydrophilic excipients. Improvements in the dissolution rate of co-grinded particles can be explained by the reduction in particle size. The use of the combination of these two methods (liquisolid and co-grinding) is in its infancy as only one article has been published so far [13]. Therefore, there is a big gap to explore the application of the combined methods on various BCS class II drugs such as celecoxib, particularly where liquisolid and grinding alone could not improve its dissolution, which is the case for celecoxib. The aim of the current study is to explore if a combination of liquisolid and grinding techniques can show a synergistic effect on the dissolution rate of celecoxib. In addition, previous studies have shown that the liquisolid technique did not significantly increase the dissolution rate of celecoxib. Therefore, in this study, the liquisolid technique was combined with co-grinding in an attempt to further improve the dissolution rate.
Materials
The following materials were used in this work: celecoxib (Darupakhsh Pharmaceuticals, Tehran), microcrystalline cellulose (Avicel PH101) (Mingtai Chemical, Taiwan), sodium starch glycolate (Mingtai Chemical, Taiwan), colloidal silicon dioxide (Mingtai Chemical, Taiwan), magnesium stearate (Mingtai Chemical, Taiwan), ethanol (Merck, Germany), polysorbate 80 (Tween 80) (Merck, Germany), polyvinylpyrrolidone (PVP-K30) (Sharlau, Belgium), polyethylene glycol 200 (Merck, Germany). Materials such as sodium chloride (Merck, Germany), monobasic potassium phosphate (Merck, Germany), sodium hydroxide (Merck, Germany), and 37% hydrochloric acid (Merck, Germany) were used to prepare simulated gastric fluid (SGF) and simulated intestinal fluid (SIF).
Preparation of Celecoxib Formulations
Preparation of Tablets Using a Combination of Ball Milling and Liquisolid Techniques
The liquid medication was prepared by dispersing celecoxib in specified amounts of polyethylene glycol 200, which was then transferred to a planetary ball mill containing several stainless steel balls (5–10 cm diameter) equipped with a 50-mL zirconia milling chamber (PM 100 RESTCH, Germany) that milled for 3 h. After uniform dispersion was achieved, PVP was slowly added to the liquid medication and mixed gently for 5 min. Next, according to the calculated load factor, a specified amount of Avicel PH101 was added and mixed with liquid medication using a mortar and pestle. Silica coating material with uniform size, which was previously sieved using a 60-mesh sieve, was then added to the mixture of silica to Avicel (with a ratio of 1:20) and was mixed for 5 min to obtain a dry and non-cohesive mixture [14,15,16].
Considering the carrier-to-coating material ratio (R), a decrease in R will increase the load factor and the weight of the final dosage form will decrease [16, 17]. However, with decreasing R-value, the dissolution rate of the drug decreases [16]. It is generally found that when R-value is 20, good drug release is observed and the weight of the final unit dosage form remains reasonable [16]; therefore, an R-value of 20 was selected for this study. Finally, sodium starch glycolate, 3.5% (w/w), was added as a super disintegrant to the obtained formulations and mixed for 15 min. Tablets containing 100 mg of celecoxib were then prepared using compression forces of 50, 100, and 150 kg/cm2 on a manual tableting machine (Riken, P-16B, Tokyo, Japan) to obtain tablets with the hardness of 5–7 kg/cm2. The formulation components are detailed in Table 1.
Preparation of Conventional Liquisolid Tablets
Conventional liquisolid tablets were prepared similarly to the combination method in the previous section, except ball milling of the liquid medication was omitted. Celecoxib and the non-volatile co-solvent (polyethylene glycol 200) were mixed using a mortar and pestle to obtain a uniform dispersion. The other excipients were then added as in the previous method, according to Table 2, and force of 50, 100, or 150 kg/cm2 was applied using a manual tableting machine to obtain tablets with a hardness of 5–7 kg/cm2.
Preparation of Direct Press Formulation (by Simple Mixing Method)
The direct compression formulation was prepared similarly to the liquisolid tablets except the non-volatile co-solvent was omitted. The tablets were compressed and prepared using a 12 mm flat-faced punch with a compression pressure of 50 kg/cm2. The composition of the formulation is detailed in Table 3.
Evaluation of Celecoxib Dissolution Rate
The dissolution apparatus recommended by the USP for tablets (USP apparatus II) was used to analyze the in vitro drug release rate of all formulations. In this method, 900 mL of SGF (pH = 1) and SIF (pH = 7.2) without enzyme were used as the dissolution media. The rotation speed of the device was set to 50 rpm. The temperature of the dissolution medium was stabilized at 37 °C during the experiment. At intervals of 5, 10, 15, 30, 45, 60, 90, and 120 min, a 5-mL sample of the medium was withdrawn from each dissolution and then replaced by 5 mL of fresh dissolution medium to maintain a constant volume. The samples were filtered using 0.45 µm Millipore syringe filters before quantification using the UV–vis absorbance at 249 nm. Preliminary data obtained from the calibration curve were used to calculate the percent drug release.
To correct for the decrease in concentration due to sampling, a correction factor was calculated using the following Eq. (1):
where Ctn is the actual concentration of the sample n, Con is the measured concentration of the sample n, v is the volume of the sample taken, V is the volume of the dissolution medium, and \(\sum_{i=1}^{n-1}\mathrm{Coi}\) is the sum of the concentrations measured for all samples before sample n.
The in vitro release profile was investigated by using similarity factor (f2) defined by the following equation:
In the equation, n is the number of the time points, and Rt and Tt are the percentage of the dissolved drug for two formulations that are being compared. The similarity factor less than 50 is considered a significant difference between the two dissolution profiles.
Friability Test
This test was carried out using 10 tablets of each formulation type. The weight was recorded then the tablets were placed inside a dual-chamber drum friability tester (Erweka) and spun for 4 min at 25 rpm. Afterward, the tablets were removed from the machine, dusted, and weighed again, and calculations were performed using the following equation:
- W 2 :
-
Final weight
- W 1 :
-
Initial weight
Acceptable friability (weight loss) for tablets under these conditions is less than 1%. The results of the friability tests are presented in Table 4.
Powder X-ray Diffraction Spectroscopy Analysis
Powder X-ray diffraction was used to investigate the solid-state properties of the prepared formulations. For this purpose, the pure drug, co-ground formulation, liquisolid formulation, physical mixture, Avicel PH101, and silica were investigated. All samples were prepared and placed on a copper plate for X-ray irradiation (Cu-k) with a wavelength of 1.5406 A. The ambient temperature was 25 °C and the 2θ angle was 5–70° with a scanning rate of 60°/min.
Differential Scanning Calorimetry
DSC thermograms of celecoxib, excipients, and formulations were obtained using a DSC-60 device (Shimadzu, Japan). The samples were weighed (3–5 mg) and placed in aluminum pans, which were pressed using a Shimadzu crimper after fitting the caps. In each test, the temperature was increased at a rate of 10 °C per minute in the range of 25–300 °C.
Investigating the Effect of Aging on the Hardness and Release Rate of the Co-ground Formulations
To investigate the effect of aging on the hardness and drug release rate of the prepared formulations and to investigate possible leakage of non-volatile co-solvent, some of the tablets from each series were selected and stored at 25 °C (away from moisture and light) for 6 months. The tablet hardness was then checked using a hardness tester (Erweka TBH 30MD) and, finally, a dissolution test was performed under the same conditions mentioned in the “Evaluation of Celecoxib Dissolution Rate” section.
Results and Discussion
Comparison of the Dissolution Profiles of the Co-ground Formulations in SGF and SIF Media
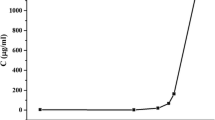
Celecoxib is a weakly acidic drug, so it is expected that the dissolution rate will increase with increasing pH. According to Fig. 1, the maximum release of the formulation that combines liquisolid and ball milling technique was 28.53% in SIF medium and 18.1% in SGF medium after 2 h (bear in mind this is a non-sink condition). There are no significant differences between the two dissolution profiles. The pKa of celecoxib is 11.1 [18] so it is non-ionized at pH below its pKa of 11.1 and the change in pH in this range does not cause a significant change in drug dissolution according to the dissolution profile shown in Fig. 1.

Dissolution profiles of LSB1 (made by combined method containing 30% API in liquid medication) in SGF and SIF media
The dissolution medium proposed by the US FDA has a pH of 12 (https://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_getalldata.cfm), which is justified by celecoxib’s pKa but is not compatible with in vivo environments. Therefore, in this study, it was preferred to use the actual pH of the stomach or small intestine. Similar results were obtained for the other formulations containing 40% and 50% API in liquid medication in both SIF and SGF.
Comparison of Dissolution Profiles of Formulations Made via Liquisolid-Ball Milled, Conventional Liquisolid, and Physical Mixture Approaches
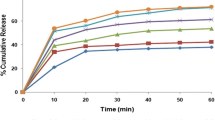
In Figs. 2, 3 and 4, it can be seen that the drug dissolution rate of the liquisolid co-ground formulation, i.e., the formulation prepared using a ball mill, is significantly faster than that of the physical mixture by a factor of about 7.5 after 60 min. As the wettability of the liquisolid formulation is improved due to the presence of hydrophilic liquid vehicle, therefore, more of the drug particles is exposed to the dissolution medium, hence improving the dissolution rate [19,20,21,22,23]. Generally, the dissolution rate of drugs in the dissolution medium depends on the contact area of the drug with the dissolution medium [24]. It is known that the specific surface area of the drug when the combination of milling and liquisolid is used is higher than celecoxib particles in DC tablets. Therefore, liquisolid co-ground formulations show a better dissolution rate compared to the other formulations. The gradient of the drug concentration versus time indicates the rate of drug dissolution [25]. At a constant rotational speed of the paddle (50 rpm) and in the same dissolution medium, the thickness of the static diffusion layer and the diffusion coefficient of the drug molecules can be considered the same. Therefore, the high dissolution rate of tablets prepared by the co-ground and classical liquisolid methods is attributed to a significant increase in the specific surface area of dispersed celecoxib [26, 27]. The Noyes–Whitney equation assumes the constant proportionality of dissolution rate to the concentration difference (Cs − C(t)) between the solubility, Cs, and the concentration of drug in the dissolution medium at time t, C(t), [28, 29]. There are other models of dissolution that are derived by extending the Noyes–Whitney equation [30,31,32,33,34,35,36,37,38,39,40]. In addition, the saturation of the drug in the microenvironment (Cs) in liquid-to-solid systems is expected to increase in liquisolid co-ground formulations. Although the small amount of liquid solvent applied to the solid in each liquisolid tablet is not sufficient to increase the overall drug solubility in the dissolution medium, it is possible that in the microenvironment, the liquid medication diffusing out of liquisolid primary particles may be sufficient to enhance the solubility of the drug by acting as a co-solvent with the aqueous dissolution medium in the diffusion boundary layer. Such an increase in Cs increases the drug dissolution rate [25, 27]. In the dissolution profiles shown in Figs. 2, 3 and 4, it can be seen that the drug release rate for the tablets made using the combination of ball milling and liquisolid method (co-ground formulation) is faster than that for the tablets made using the classical liquisolid method. This is owing to the influence of ball milling of the celecoxib particles, resulting in a reduction in the particle size, which improves the drug wettability with the non-volatile co-solvent [6, 19, 41,42,43,44]. Moreover, according to the Noyes–Whitney equation, increased solubility is expected with a decrease in drug particle size.

Release profiles of the prepared formulations (30%) in SIF medium

Release profiles of the prepared formulations (40%) in SIF medium

Release profiles of the prepared formulations (50%) in SIF medium

Diffractograms of formulations made by direct compression, classic liquisolid and combined methods, pure drug, and Avicel
Figures 2, 3 and 4 show that the combination of liquisolid with ball milling and liquisolid only formulations has a faster drug release rate than the physical mixture. The tablets prepared with the combined method have a better dissolution profile than the tablets prepared with the classical liquisolid method. Furthermore, when the concentration of the drug in the liquid medication is increased, the dissolution rate of the drug decreases. This difference can be explained by the theory of solubilization and molecular dispersion state of the drug in the formulations, as discussed earlier. By using a higher amount of solvent in the formulation, higher molecular dispersion of the drug is achieved, leading to an increased release rate for the tablets prepared using 30% API in the liquid medication compared to those containing 40% and 50%.
Powder X-ray Diffraction Analysis
The polymorphic structure of a drug affects its dissolution rate and bioavailability, so the polymorphic changes to celecoxib during the process should be considered. The diffractograms of celecoxib, Avicel, silica, and the formulation prepared using different methods are shown in Fig. 5. The crystalline celecoxib has sharp peaks at 5°, 10.5°, 16°, and 21.5°. This celecoxib diffractogram is consistent with reports in the literature [32]. The liquisolid and physical mixture formulations showed similar diffraction patterns, except that the peak heights were significantly lower. This is due to the lower concentration of the drug in these systems compared to others and also a possible reduction in the degree of crystallinity. In addition, no peaks other than those related to pure celecoxib and Avicel were observed in the case of the liquisolid systems, indicating no chemical interaction between the drug and the excipients.

Thermograms of different formulations containing 50% celecoxib and excipients
DSC Thermogram Analysis
In the thermogram of pure celecoxib, the melting peak is observed at 161. 26 °C, which is shown by the sharp endothermic peak. According to Fig. 6, in the physical mixture thermogram, a small peak is also observed at this point, which is due to the low concentration of the drug in the formulation. However, there is no peak in the co-ground and classic liquisolid formulations, which could be due to changes in the degree of crystallinity and amorphous form of the drug.

Dissolution profiles for freshly prepared tablets containing 40% celecoxib made with the combined liquisolid and ball milling technique with the corresponding aged tablet (6 months) in SIF medium
Evaluation of the Effect of Aging on the Hardness and Drug Dissolution Rate
The liquisolid tablets use a non-volatile co-solvent within the matrix of the tablet, so over time this solvent may leak out to the surface of the tablet and, in addition to disrupting the structure of the dosage form, it may cause the tablets to adhere to each other. In addition, it can change the hardness of the tablets over time, all of which can affect the release pattern and stability of the drug. To investigate the effect of aging on the hardness and release rate of the co-ground formulations and to investigate possible leakage of liquid solvent, many tablets from the 40% co-ground formulation series were selected and stored at room temperature (away from moisture and light) for 6 months. The hardness was then checked using a hardness tester and, finally, a dissolution test was performed with the same conditions as mentioned before. No visible cracks or leaks were observed in the tablets. The results of the hardness test showed that there was no significant difference between the initial hardness and hardness of tablets after 6 months (6–7 kg/cm2). It can be concluded that the hardness of the prepared liquisolid tablets does not change significantly during storage and solvent leakage does not occur on the tablet surface. The dissolution profiles of the freshly prepared and aged tablets are shown in Figs. 7 and 8. As can be seen, there was no significant change in the dissolution profile of freshly prepared tablets and aged tablets in the SIF and SGF environments (f2 > 50).

Dissolution profiles for freshly prepared tablets containing 40% celecoxib made with the combined liquisolid and ball milling technique with the corresponding aged tablet (6 months) in SGF medium
Conclusion
The data in the study showed that the liquisolid methods can be used to enhance the dissolution rate of water-insoluble drugs such as celecoxib. Compared to DC tablets, liquisolid tablets have a significantly higher in vitro drug dissolution rate. This enhanced drug dissolution may be due to the increased wettability and the contact area between the drug and the dissolution medium. In comparing the classic liquisolid method with the combination method (liquisolid combined with co-ground technique), the latter method exhibited a higher dissolution rate, which was attributed to the increased wettability and smaller particle size. Therefore, this method can be used to increase the dissolution rate for drugs that have a relatively low rate of dissolution. The results from the solid-state study showed that in the solid and liquisolid formulations, no additional peaks or peak displacement were observed; only the intensity of the drug peaks decreased due to the lower degree of crystallinity of the drug in these systems. Aging did not affect the hardness or release profile of the tablets obtained by the combination method. Therefore, this method can be used to improve the performance of the drug.
Availability of Data and Material
The datasets generated during this work can be available upon request.
References
Amidon GL, et al. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20.
Löbenberg R, Amidon GL. Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur J Pharm Biopharm. 2000;50(1):3–12.
Ohm A. Interaction of Bay t 3839 coprecipitates with insoluble excipients. Eur J Pharm Biopharm. 2000;49(2):183–9.
Morgen M, et al. Polymeric nanoparticles for increased oral bioavailability and rapid absorption using celecoxib as a model of a low-solubility, high-permeability drug. Pharm Res. 2012;29(2):427–40.
Sobhani T, Shokri J, Javadzadeh Y. Enhancement of fingolimod content uniformity as a low-dose drug with narrow therapeutic index using liquisolid technique. J Pharm Innov. 2020;1–11.
Javadzadeh Y, Musaalrezaei L, Nokhodchi A. Liquisolid technique as a new approach to sustain propranolol hydrochloride release from tablet matrices. Int J Pharm. 2008;362(1–2):102–8.
Muselík J, et al. Influence of process parameters on content uniformity of a low dose active pharmaceutical ingredient in a tablet formulation according to GMP. Acta Pharm. 2014;64(3):355–67.
Tong Y, et al. Systematic development of self-nanoemulsifying liquisolid tablets to improve the dissolution and oral bioavailability of an oily drug, vitamin K1. Pharmaceutics. 2018;10(3):96.
Pathak A, et al. A review on liquisolid technology. World J Pharma Res. 2012;1(3):500–12.
Bhavya E, Dhere M. Liquisolid compacts technique of poor water soluble drugs: an overview. Res J Pharm Technol. 2021;14(10):5569–72.
Jaipakdee N, Tabboon P, Limpongsa E. Application of a liquisolid technique to cannabis sativa extract compacts: effect of liquid vehicles on the dissolution enhancement and stability of cannabinoids. Int J Pharm 2022;612:121277.
Javadzadeh Y, Jafari-Navimipour B, Nokhodchi A. Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine). Int J Pharm. 2007;341(1–2):26–34.
Azharshekoufeh L, et al. Liquigroud technique: a new concept for enhancing dissolution rate of glibenclamide by combination of liquisolid and co-grinding technologies. BioImpacts. 2017;7(1):5.
Riippi M, et al. The effect of compression force on surface structure, crushing strength, friability and disintegration time of erythromycin acistrate tablets. Eur J Pharm Biopharm. 1998;46(3):339–45.
Summers MP, Enever RP. Preparation and properties of solid dispersion system containing citric acid and primidone. J Pharm Sci. 1976;65(11):1613–7.
Javadzadeh Y, et al. Enhancement of dissolution rate of piroxicam using liquisolid compacts. Farmaco. 2005;60(4):361–5.
Spireas S, Wang T, Grover R. Effect of powder substrate on the dissolution properties of methyclothiazide liquisolid compacts. Drug Dev Ind Pharm. 1999;25(2):163–8.
Ha ES, et al. Formulation, characterization, and in vivo evaluation of celecoxib-PVP solid dispersion nanoparticles using supercritical antisolvent process. Molecules. 2014;19(12):20325–39.
Elkordy AA, Tan XN, Essa EA. Spironolactone release from liquisolid formulations prepared with Capryol™ 90, Solutol® HS-15 and Kollicoat® SR 30 D as non-volatile liquid vehicles. Eur J Pharm Biopharm. 2013;83(2):203–23.
Khames A. Liquisolid technique: A promising alternative to conventional coating for improvement of drug photostability in solid dosage forms. Expert Opin Drug Deliv. 2013;10(10):1335–43.
Gubbi SR, Jarag R. Formulation and characterization of atorvastatin calcium liquisolid compacts. Asian J Pharm Sci. 2010;5(2):50–60.
Gonjari ID, Karmarkar AB, Hosmani AH. Evaluation of in vitro dissolution profile comparison methods of sustained release tramadol hydrochloride liquisolid compact formulations with marketed sustained release tablets. Dig J Nanomater Biostruct. 2009;4(4):651–61.
Burra S, Yamsani M, Vobalaboina V. The liquisolid technique: an overview. Braz J Pharm Sci. 2011;47(3):475–82.
Dizaj SM, et al. Nanosizing of drugs: effect on dissolution rate. Research in pharmaceutical sciences. 2015;10(2):95.
Wadke DA, Serajuddin AT, Jacobson H. Preformulation testing Pharmaceutical dosage forms: Tablets. 1989;1:1–73.
Spireas S, Sadu S. Enhancement of prednisolone dissolution properties using liquisolid compacts. Int J Pharm. 1998;166(2):177–88.
Bertagnolli MM, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355(9):873–84.
Noyes AA, Whitney WB. Über die Auflösungsgeschwindigkeit von festen Stoffen in ihren eigenen Lösungen. Z Phys Chem. 1897;23(1):689–92.
Macheras P. Biopharmaceutics of orally administered drugs. CRC Press. 1995.
Adams E, et al. Application of linear mixed effects models to the evaluation of dissolution profiles. Int J Pharm. 2001;226(1–2):107–25.
Adams E, et al. Non-linear mixed effects models for the evaluation of dissolution profiles. Int J Pharm. 2002;240(1–2):37–53.
Homayouni A, et al. Preparation and characterization of celecoxib solid dispersions; comparison of poloxamer-188 and PVP-K30 as carriers. Iran J Basic Med Sci. 2014;17(5):322–31.
Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci 2001;13(2):123–133.
Frutos G, de Villa MR. Nonlinear mixed-effects model for the dissolution assays of drugs. J Control Release. 2004;94(2–3):381–389.
Hintz RJ, Johnson KC. The effect of particle size distribution on dissolution rate and oral absorption. Int J Pharm. 1989;51(1):9–17.
Koizumi T, Ritthidej GC, Phaechamud T. Mechanistic modeling of drug release from chitosan coated tablets. J Control Release. 2001;70(3):277–84.
Macheras P, Dokoumetzidis A. On the heterogeneity of drug dissolution and release. Pharm Res. 2000;17(2):108–12.
Sathe PM, Tsong Y, Shah VP. In-vitro dissolution profile comparison: statistics and analysis, model dependent approach. Pharm Res. 1996;13(12):1799–803.
Polli JE, et al. Methods to compare dissolution profiles and a rationale for wide dissolution specifications for metoprolol tartrate tablets. J Pharm Sci. 1997;86(6):690–700.
Wang J, Flanagan DR. General solution for diffusion-controlled dissolution of spherical particles. 1. Theory. J Pharma Sci. 1999;88(7):731–738.
Suliman AS, Anderson RJ, Elkordy AA. Norfloxacin as a model hydrophobic drug with unique release from liquisolid formulations prepared with PEG200 and Synperonic PE/L-61 non-volatile liquid vehicles. Powder Technol. 2014;257:156–67.
Fahmy RH, Kassem MA. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In vitro and in vivo evaluation. Eur J Pharm Biopharm. 2008;69(3):993–1003.
Javadzadeh Y, et al. An investigation of physicochemical properties of piroxicam liquisolid compacts. Pharm Dev Technol. 2007;12(3):337–43.
Lu M, et al. Dissolution enhancement of tadalafil by liquisolid technique. Pharm Dev Technol. 2017;22(1):77–89.
Funding
This study was financially supported by the National Institute for Medical Research Development (NIMAD) (grant number: 977080).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable.
Consent for Publication
All authors are happy with this publication.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nazem, N.M., Shokri, J., Nourani, N. et al. Combining Liquisolid and Co-grinding Techniques to Enhance the Dissolution Rate of Celecoxib. J Pharm Innov 18, 300–309 (2023). https://doi.org/10.1007/s12247-022-09641-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12247-022-09641-1