
Abstract
Purpose
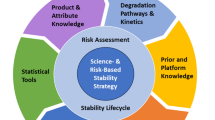
Lean stability is a science- and risk-based initiative which utilizes the enhanced understanding of drug substance and drug product physical and chemical characteristics to (1) reduce and optimize the design of standard stability protocols; (2) expedite the generation of stability data without impact to safety, efficacy, or quality of the product; and (3) decrease time to market for innovative drugs. Lean stability was introduced in the early 2000s [ICH: Guideline Q1A(R2) (2003), ICH: Guideline Q1D (2002)] followed by reduced stability protocols, focusing on the critical quality attributes and critical time points, being reported in the literature [Skrdla et al. (J Pharm Biomed Anal 50: 794–796, 2009)]. While the concept of lean testing is not entirely new, and it is currently a part of several regulatory guidances, it continues to evolve and gain acceptance of the industry and regulators.
Methods
In this review, twelve case studies are presented where stability data was collected during clinical, registration, and post-approval phases of the product development.
Results
Case studies summarize the lean stability testing design, the strategies applied during the regulatory filing and the outcomes of the regulatory filings.
Conclusion
The authors expect that the case studies presented in this review will increase the visibility of lean stability, facilitate overcoming of the existing challenges, and accelerate the global regulatory acceptance of lean stability practices in the industry.
Similar content being viewed by others

Introduction
This manuscript is intended to encourage risk-based decision making in stability testing and to further drive for implementation of lean stability strategies through delineation of case studies from recently accepted practices. Lean stability is a science- and risk-based approach that leverages enhanced knowledge about the stability behavior of a product to focus stability programs on the highest risk attributes and time points. Application of lean stability concepts enables more efficient, product-specific stability studies and is fully aligned with modern approaches toward product and process development [1], leading to a reduced protocol compared to a standard, ICH Q1A-type program. The concept of lean stability has been applied for over two decades, beginning with the ICH (Q1A(R2), Q1D) guidance allowing for bracketing and matrixing during stability testing [2], and continuing into later ICH (Q8–12) and WHO guidances stipulating the acceptability of scientifically driven approaches towards meeting stability requirements [1, 3,4,5,6,7]. The concept of reducing the testing plan to include only critical attributes that define the product’s stability and performance over time have been previously published [8,9,10]. While these approaches have primarily had positive regulatory responses [11], they have not been included in regulations to date.
In 2016, the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ) launched the lean stability working group focused on increasing the understanding and acceptance of science- and risk-based protocol strategies. The working group has approximately 20 members from 14 companies across the pharmaceutical industry who have contributed the case studies contained in this paper. The companies represent development and manufacture of multiple dosage forms (e.g., solid oral, parenteral, combination products) and support stability strategies throughout the product life cycle (i.e. drug substances, drug products) across pharmaceutical product modalities (e.g., small molecules, biologics, etc.). These case studies demonstrate how industry has successfully expanded beyond the strategies initially defined in the ICH Q1 guidance as well as provide insight towards how to continually improve efficiency within stability studies and product understanding throughout the product development life cycle: clinical development, registration phase, and post-approval. The lean stability strategy is dependent upon a robust understanding of the product design and/or profile, and considers existing data from development or accelerated studies, risk-based predictive stability, and literature.
Industry spends unnecessary resources initiating and maintaining long-term stability protocols that include non-stability indicating tests and non-value-added time points throughout the product’s life cycle. Implementing lean stability strategies requires the adjustment of stability protocols to improve efficiency and expedite results without impacting safety, efficacy, or quality of the product. Lean strategies are applicable at all stages of the product life cycle from development to marketed phase and to all pharmaceutical product types and are allowable by ICH Q1A (R2) [12] as “Alternative approaches can be used when there are scientifically justifiable reasons.” In addition, these strategies do not reduce product stability risk knowledge but rather could benefit the patient through decreasing the time to market for new medicines.
An example of a lean stability approach used in the post approval phase is shown below. A lean stability approach focuses on using stability indicating attributes and existing stability knowledge of a drug substance or drug product to focus on the right tests, time points, and conditions throughout the study. Where the overall stability is well-understood and the drug substance or drug product stability attributes are predictable, a lean approach is warranted. Table 1 provides an example of a standard full stability protocol and outlines typical testing attributes for a drug product. While there are many situations where lean stability may be applied, Table 2 shows an example lean stability protocol approach for a stable room temperature drug product where conditions, time points, and testing attributes have been reduced yet still provides adequate stability information. This approach would result in cost and time savings without impacting quality and safety of the product. This is just one of many examples; conditions and time points may be selected based on specific product attributes and intended region of market. For example, testing at the 30 °C/75% RH condition, as opposed to 30 °C/65% RH, to allow for global product development may not be standard practice at all companies.
This example could also apply to setting a shelf-life for a stable product in the clinical phase of development. With sufficient prior knowledge, this lean approach example, and the addition of an accelerated condition (40 °C/75% RH), the proposed shelf life dating could be supported based upon results for the stability—indicating test attributes and any supporting data that may be available. Supporting data could consist of existing standard stability data and/or information from accelerated stability approaches (risk-based predictive stability) [13,14,15] where the accelerated data can be statistically analyzed (e.g., ASM, ASAP, etc.)Footnote 1 [15] to model shelf-life limiting attributes (SLLAs). It is important to note that the lean stability approach may differ from that in Table 2, customized based on product-specific understanding obtained from prior stability studies of critical quality attribute(s) and/or shelf-life limiting attribute(s).
The risks to implementing such an approach include the potential non-acceptance or queries from health authorities related to less data being collected than a standard stability protocol. In order to remediate these risks, thorough risk assessments should be completed for each product to understand the reliability of methods and uncertainty of design by leveraging previously gained knowledge.
Barriers to implementing lean strategies have been experienced both internally and externally to companies within the pharmaceutical industry. The most significant hurdle colleagues have faced when proposing lean stability strategies is internal company conservatism within several departments and management levels. External uncertainties such as a lack of alignment regarding regulatory positions have been reported, both between agencies and within a single agency. Efforts to influence and better understand concerns of regulatory agencies should be made. By presenting case studies collected from multiple companies, the authors hope to provide an improved understanding of lean stability approaches and the benefit they can provide to product development.
Case Studies
A lean stability strategy can take many forms. It should be product specific, dependent on the stage of development and reflect the product knowledge available at the time. In the clinical development phase, while development is under way and changes and improvements are constant, this may take the form of utilizing predictive tools and/or confirmatory studies to demonstrate a change does not impact stability rather than re-initiating a long-term stability protocol, to justify re-test period or shelf-life, or to identify which tests are stability indicating. In the registration phase, a lean strategy may leverage the cumulative clinical and registration stability data to justify reducing the tests, time points and/or storage conditions that are necessary to monitor stability on an annual basis. In the post-approval phase, product knowledge and stability understanding are highest and there are many opportunities to leverage lean strategies to support a change. Following are examples of case studies collected during each phase of development: clinical development, registration, and post-approval. Table 3 provides an overview of the presented case studies.
Clinical Development
Case Study 1
In this case study, the drug substance stability data were used to support drug in capsule (DiC) product. The scope of the project spanned across 5 small molecules with drug in capsule formulations. The FDA Guidance for Industry, cGMP for Phase 1 Investigational Drugs (U.S. Food and Drug Administration, 2008) allows representative samples of phase 1 investigational drugs to be used to monitor stability and quality. In the case of DiC formulations it was determined that the drug substance stability data was representative of the DiC stability. The justification in the Clinical Trial Application (CTA) included referencing the Common Technical Document (CTD) Section S.7 in Section P.8. The CTA also included reference to accelerated stability data on the drug substance as part of the justification. Regulatory submissions were performed in the US, France and Spain. Challenges were received in two cases (US and Spain) and the agencies requested that the DiC be placed on long-term stability concurrent with the clinical study. Due to timing considerations, further technical discussion was not pursued, and the DiC was placed on long-term stability. The primary benefit was still realized by this approach; time savings in not waiting for the one-month drug product stability data prior to IND or CTA filing.
The following wording was included in Section P.8 of a CTA as justification for not placing a DiC on stability, following a risk assessment to ensure the drug substance is not susceptible to increased moisture uptake by the hydroxypropylmethylcellulose (HPMC) capsule shells.
Summary of Stability Studies
Representative batches of drug product will not be formally evaluated for stability. The drug substance stability (Section S.7) will be monitored to assure pharmaceutical performance and strength for the duration of clinical trials.
The drug product is a simple drug in capsule using hydroxypropylmethylcellulose (HPMC) capsule shells (see Section P.3.3) and contains no additional excipients. In addition to the ongoing stability study (Section S.7), the general stability of the drug substance is supported by solid state degradation studies. After exposure to high temperatures/high humidity (70 °C/75% RH), and light conditions, the purity of the material was unchanged.
Since the drug substance does not exhibit any physical or chemical characteristics that could impact drug in capsule stability (e.g., hydrolytic instability), stability studies of drug in capsule product are not necessary to ensure safety and quality of the clinical trial materials. The stability of the drug substance is representative of the stability of the drug in capsule formulation.
Case Study 2
In this case study, accelerated short-term stability comparison studies were leveraged for subsequent drug substance campaigns with minimal synthetic route changes. An internal risk assessment was performed to determine the potential impact of the synthetic route changes. This assessment was then used to inform the comparative stability study. The study design consisted of a short term, typically 2 weeks to 1 month, accelerated stability study to establish comparability of a new batch of drug substance back to the original drug substance batch. Accelerated stability conditions should be based on prior degradation chemistry knowledge, but typical conditions range from 40 °C/75%RH to 70 °C/75%RH for stable drug substance batches.
The comparability between batches was used as justification in Section S.7 of the CTA for not placing subsequent drug substance batches on long term stability. The approach has been used to support regulatory filings in the US and EU countries, with no questions received during the approval process.
The following wording was included in the CTA to justify the accelerated comparison study.
Batches Drug substance-1 and Drug substance-2 were subjected to accelerated stability conditions (70 °C/75% RH both open dish and closed) through four weeks. The results of this study support the comparability of these lots with respect to stability. Based on the similarity of the synthetic routes and the comparable stability profiles under accelerated conditions, the stability of Batch Drug substance-2 is considered representative of Batch Drug substance-1. No long-term formal stability studies of Batch Drug substance-2 are planned.
Case Study 3
An approach used to support many drug substance in bottle regulatory filings is provision of 3 weeks of drug substance stability data at 70 °C/75%RH in the initial clinical submission to justify a 15-month drug substance retest period [16]. This data may also support a 12-month Powder for Oral Solution (PfOS) clinical shelf life (PfOS consists of drug substance in a bottle with no additional excipients). This lean stability strategy includes a commitment to complete long-term drug substance stability studies for the duration of the clinical study. The approach has been successfully approved by both the US and Germany.
Case Study 4
Exclusion of assay testing has been routinely proposed in CTAs in situations where the drug substance stability batch is the same as the reference standard batch [17]. Justification for this approach argues that since the stability batch and reference standard are stored under the same conditions, assay results would only reflect method accuracy rather than degradation. Although not all countries required stability sections in the CTA, the approach was leveraged to support submissions in the following countries: US, Canada, Hungary, Slovakia, Romania, Bulgaria, India, Philippines, Taiwan, and Turkey. This approach resulted in regulatory queries but was ultimately accepted in all countries except for Canada. For two separate submissions, Belgium asked to include this test in future submissions for a specific product. Czech Republic also responded with a query, which was successfully addressed by providing assay using mass balance approach with the commitment to set up a future lot on stability and monitor assay. Canada, however, has not accepted this approach.
Registration Phase
Case Study 5
A registration strategy for drug product primary stability was developed to support seven different dosage strengths (compositionally proportional) where each dosage had two packaging configurations. An ICH Q1D bracketing design was proposed to support the NDA submission and was agreed to during a pre-NDA type C meeting with US FDA. The primary stability strategy included testing on three batches representing the lowest and the highest strengths in both packaging configurations (total of 12 batches). Supportive stability testing was performed on one batch for each of the five intermediate dosage strengths for each packaging configuration. The post-approval stability protocol to confirm expiry included in the NDA submission indicated that formal stability studies (e.g., the ICH Q1A stability commitment batches) could be performed on three batches of the lowest and 3 batches of the highest strength in each packaging configuration.
Case Study 6
A proposal to use a bracketing design according to ICH Q1D for an NDA submission was agreed to by US FDA at an End of Phase 2 type B meeting. This program had five different dosage strengths with the same (compositionally proportional) formulation and packaging configuration. The primary stability to support registration was performed on more than three batches of each of the lowest and the highest dosage strengths. Supportive stability testing was performed on two batches of one intermediate dosage strength. The post approval stability protocol included in the NDA submission showed a bracketing design and the stability commitment included 3 batches of the lowest and highest dosage strengths. This approach resulted in a significant amount of resource savings while providing a sufficient stability data package to evaluate the quality and safety of the product.
Case Study 7
An NDA was submitted for a complex modified release (MR) product with multiple strengths in capsules manufactured from common pellets. The primary stability data supported an initial shelf life of 36 months. The NDA did not include a protocol commitment to confirm expiry, since the primary stability batches were manufactured at the commercial site and commercial scale.
For the annual batch commitment, a reduced protocol was proposed relative to the primary stability protocol. The protocol proposed to test one batch of any strength per year. This was considered justified since stability data demonstrated that the formulation provided equivalent performance with comparative dissolution profiles for all strengths. Further, release and stability data were comparable across all strengths. Assay was monitored across all strengths with no strength related trends observed for any quality attribute. The annual batch commitment also proposed removal of assay testing and water content testing since these tests were demonstrated to not be stability indicating. The annual protocol also proposed long-term conditions (25 °C/60% RH) only and a reduction of time points to annual testing. The reductions to storage conditions (long-term only) and time points were accepted, but the other reduction proposals listed above were not accepted. Regarding the proposal to only test one batch of any strength per year, the FDA indicated that additional strengths should be tested when manufactured from different intermediate pellet batches. Assay and water were also added as tests in response to FDA queries. For assay, the FDA indicated that after sufficient product history is established, a prior approval supplement could be submitted to request the removal of the assay requirement from stability testing. For water testing, the sponsor had proposed that neither release nor stability criteria were required to ensure product quality. The FDA indicated that water content of the drug product should be monitored as part of release and stability testing. The sponsor agreed that after additional data were accrued, either an acceptance criterion for water content would be proposed in a post-approval CBE-30 supplement or justification of non-inclusion for the water test would be provided via a general correspondence.
Case Study 8
A stable, well-understood drug product (IR (Immediate Release) capsule) was filed with a standard primary stability protocol and data to support the initial shelf-life claim. An alternate proposal was made for the post-approval stability protocol. To limit wasted supplies for a low-volume commercial program, the applicant proposed to utilize non-printed capsules for 2 of the 3 lots for 2 of the 3 strengths. This approach enabled the capsules that were not used for stability studies to be used for blinded clinical studies. This strategy was filed and accepted globally.
The commitment to conduct annual batch stability proposed significant reductions relative to the primary stability protocol. The approved protocol included only annual time points at the long-term condition (30 °C/75%RH, used to fulfill global requirements) and reduced the quality attribute testing to appearance, assay, dissolution, and degradation products.
Post-approval
Case Study 9
In this case study, a lean stability strategy was applied for post approval/annual stability batches of a small molecule oral drug substance and drug product. Only the long-term storage condition was required, and no data was proposed to be collected for the accelerated storage condition. Description, assay, impurities/degradation products, dissolution (for drug product only), and LOD (for DS only) were the required tests. A strategy to reduce time points relative to the standard ICH registration phase was also applied; the 3 M and 9 M timepoints were removed resulting in a stability schedule consisting only of initial, 6 M, 12 M, 18 M, 24 M, and annually thereafter if longer dating would be pursued. Recent submissions occurred in the following countries: EU, Japan, US, Canada, Switzerland, Australia, Taiwan, South Korea, Hong Kong, Brazil, India, Turkey, Russia, Mexico, UAE, Argentina, and Kuwait. No regulatory questions have been received to date. The submissions required no added justification in CTD section 3.2.P.8.2.
Case Study 10
This case study relates to a large product family in semi-permeable containers, spanning multiple US regulatory filings, separated into multiple categories based on formulation, container type and filling volume. The historical requirement related to the annual commitment batch testing was to perform stability testing on one (1) formulation/container combination per category. As there were multiple categories in the family, this approach resulted in multiple batches of product being placed on stability annually.
A lean stability strategy was proposed which included the following elements:
-
Proposal 1: Revision to product categorization based on review of historical data supporting: robust stability profiles, stability trends consistent across products, demonstrated understanding of the SLLA. The revised categorization ensured testing was performed for each regulatory filing annually, while significantly reducing the number of batches required.
-
Proposal 2: Reduction in number of time points tested—less than ICH recommended [12].
-
Proposal 3: Reduction in testing within a time point—some quality attributes have been demonstrated to not trend/change over time or are stable solutes. Testing at multiple time points did not add valuable information to the stability dataset.
The regulatory strategy consisted of providing a summary of the proposal, with technical rationale for each element of the change. Additionally, it was noted in the regulatory supplement that this product family had significant history supporting robustness of the product performance. The lean stability proposal was approved successfully with no questions. The overall benefit of the initiative was a reduction of approximately 18 batches placed into the stability program annually.
Case Study 11
This case study covers the submission of a registration stability package for a biologic in accordance with the recommended batch enrollment and timepoint frequency prescribed by ICH Q5C [18] and ICH Q1D [2]. The stability data provided the foundation for approvals of reduced testing in post approval stability monitoring commitments from several agencies.
The marketing authorization applications contained one drug substance presentation (long-term storage of less than or equal to − 60 °C) stored in rigid polymeric bottles and three strengths of a refrigerated liquid drug product presentation filled into the same prefilled syringe container closure system. The registration package stability data set included five drug substance batches representative of the commercial manufacturing process, with 60 months of long-term storage data submitted for one batch and 36 months of long-term storage data submitted for the other batches. Six months of stability data was submitted for four drug substance batches representative of the commercial process at the following accelerated and stressed storage conditions of 5 °C and 25 °C/60%RH, respectively. For drug product, a bracketing approach was used in accordance with ICH Q1D [2]: at least three representative lots at both the minimum and maximum strengths intended for marketing were enrolled in the stability program, together with one representative lot at the intermediate strength. Six months of data at the accelerated storage condition of 25 °C/60%RH were obtained for all enrolled lots. Long-term stability monitoring for all the registration stability drug substance and drug product batches was conducted at the intervals prescribed by ICH Q5C [18]. The 25 °C/60%RH storage condition demonstrated that three of the analytical methods were stability-indicating, which enabled an evaluation of rates of degradation across the drug product strength range in accordance with ICH Q1E [19].
Based on the consistency of the stability profile among the registration batches, reduced testing approaches were proposed to agencies in the post approval commitment for stability monitoring of batches enrolled annually beyond the registration batches. Due to the absence of change in quality attributes in drug substance stored at the long-term condition, the sponsor proposed to only monitor at this storage temperature annually up to the proposed expiry. For drug product, the sponsor proposed to monitor annual enrollment drug product lots at only the long-term condition through the end of shelf life. The sponsor also proposed enrollment of only one lot of one of the three product strengths per year into the stability program and omitting the 3-month and 9-month stability intervals as prescribed by ICH Q5C [18].
The reduced drug substance stability testing post approval commitment was approved by all agencies with which the S.7.2 Section was registered. This lean stability approach enabled an annual QC workload savings of 4 drug substance stability time points per year relative to the intervals recommended for registration studies recommended by ICH Q5C [18].
As for drug product, no objections were received to the long-term storage drug product post approval stability monitoring commitment proposed in the P.8.2 Section. However, the FDA specified that monitoring each enrolled lot for 6 months at the accelerated condition of 25 °C/60%RH was also required to receive marketing authorization, to provide continued assurance of the consistency of the stability profile across drug product strengths. Nevertheless, this lean stability approach enabled an annual QC workload savings of 22 drug product stability time points per year relative to ICH Q5C [18] and relative to enrolling annually into the stability program one lot of every marketed drug product strength according to the storage conditions and testing frequency registered in Section P.8.2.
Case Study 12
A science- and risk-based stability strategy was developed to support a drug substance manufacturing site change for a marketed small molecule solid oral dosage form product. There were no changes to the drug substance manufacturing process associated with the site change, and there were no proposed changes to the drug product. The drug substance and drug product are very stable with no identified SLLAs. Globally, the product is approved in 79 markets. To change the drug substance manufacturing site, 23 of those countries required some form of supporting stability data. Six of those required either 6 months or 12 months of drug substance stability data from the new drug substance manufacturing site to support the change. Seventeen countries required drug substance and drug product stability data to support the change. Given the high product understanding and stability knowledge, a justification was established to support a reduced stability protocol, both for the drug substance stability requirements and for the drug product stability requirements. This strategy was used for all but two countries that require stability data.
For the science and risk-based protocol, time points and tests were reduced. For both drug substance and drug product, reduced time points included 3 months and 6 months at 40 °C/75%RH and only annual time points at 30 °C/75%RH. Reduced testing for drug substance included only appearance and purity and reduced testing for drug product included only appearance, assay, degradation product, and dissolution testing.
Within the six countries that require drug substance stability data to support the change, five countries received the reduced protocol. Four of those are now approved and one remains under review. The sixth country required a more extensive drug substance protocol that included additional time points and assay testing. From the perspective of the initial registration stability protocol, this protocol was also significantly reduced with respect to the quality attributes that were assessed. This approach was also approved.
Within the remaining countries (23) that require some level of drug substance data from the new manufacturing site and drug product stability data on batches manufactured using drug substance from the proposed site, 12 submissions were filed with the science and risk-based protocol. The reduced protocol strategy will be submitted in four additional countries (not yet submitted) and a more standard stability strategy will be submitted in one additional country. Of the 12 countries where the reduced protocols were submitted, five were approved and seven are under review. The country where a more standard stability strategy was submitted included additional time points and assay testing for drug substance and additional time points for drug product. The quality attribute testing for drug product remained the same as the reduced protocol.
Conclusions
The case studies presented herein delineate how the combination of scientific rationale and product knowledge have been leveraged successfully to develop stability strategies that were robust and efficient. It is evident that there are a multitude of opportunities for scientifically sound lean stability approaches to be adopted. While hurdles towards the application of lean approaches remain, both externally from regulatory authorities and potentially more so internally within companies, there are many scientifically sound lean approaches that could be widely applied without significant regulatory challenge. Determining such approaches relies upon identifying the tests that are stability indicating (stability-related quality attributes or SRQAs) and shelf-life limiting attributes (SLLAs) as well as understanding the overall chemical and physical stability of a compound or drug product. Tests which can be deemed non-stability indicating can be removed from or reduced within the stability protocol, and where a robust data package is available, bracketing approaches are often used. One common example of a lean testing approach might be elimination of water testing for drug product where moisture content is not found to be a SLLA. Similarly, microbial testing when initial sterility and container closure integrity are demonstrated represents a potential opportunity for technically justifying removal from or reduction of this testing in the stability protocol. The reduction of time points and bracketing formulations are additional common examples found in many of the case studies shared.
Although lean stability concepts have been discussed within ICH since the early 2000s with the advent of bracketing and matrixing approaches, a unified regulatory response to such scientifically driven practices has not yet been achieved. Without global acceptance of lean stability strategies, many companies performing global development will inevitably default to the most conservative parameters in order to leverage the same dataset to fulfill the expectations of all countries, although the same level of detail might not be part of regional specific submissions. Therefore, the primary goal of this publication is to further drive for implementation through increasing exposure to accepted practices of lean stability utilized throughout the development continuum. Success in this endeavor will ultimately benefit the patient by fostering an increased efficiency in the delivery of innovative medicines.
Data Availability
All case study information provided has been approved by the contributing company/author.
Notes
Accelerated stability modeling (ASM) and accelerated stability assessment program (ASAP) are specific approaches for performing science- and risk-based predictive stability assessments. Science- and risk-based predictive stability is defined as short-term stability studies using high temperatures (and varying humidities) and modeling to predict stability at temperatures (and defined humidities) intended for long-term storage conditions.
References
ICH: Guideline Q8(R2) (2009) pharmaceutical development, Step 4 version. https://database.ich.org/sites/default/files/Q1A%28R2%29%20Step4.pdf
ICH: Guideline Q1D (2002) Bracketing and matrixing designs for stability testing of new drug substances and products, Step 4 version. https://database.ich.org/sites/default/files/Q1D_Guideline.pdf
ICH: Guideline Q9 (2005) Quality risk management, Step 4 version. https://database.ich.org/sites/default/files/Q9_Guideline.pdf
ICH: Guideline Q10 (2008) Pharmaceutical quality system, Step 4 version. https://database.ich.org/sites/default/files/Q10_Guideline.pdf
ICH: Guideline Q11 (2012) Development and manufacture of drug substances (chemical entities and biotechnological/biological entities, Step 4 version. https://database.ich.org/sites/default/files/Q10_Guideline.pdf
ICH: Guideline Q12 (2019) Technical and regulatory considerations for pharmaceutical product lifecycle management, Step 4 version. https://database.ich.org/sites/default/files/Q12_Guideline_Step4_2019_1119.pdf
World Health Organization (2018) Annex 10 Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. https://database.ich.org/sites/default/files/Q1F_Stability_Guideline_WHO_2018.pdf
Skrdla PJ, Wang T, Antonucci V, Dowling T, Ge Z, Ellison D, Curran J, Mohan G, Wyyratt J. Use of a quality-by-design approach to justify removal of the HPLC weight % assay from routine stability testing protocols. J Pharm Biomed Anal ;2009 50: 794–796.
Colgan ST, Watson TJ, Whipple RD, Nosal R, Beaman JV, Antonis DMD. The application of science-and risk-based concepts to drug substance stability strategies. J Pharm Innov. 2012;7(3–4):205–13.
Colgan ST, Timpano RJ, Roberts M, Weaver R, Ryan K, Fields KW, et al. Opportunities for lean stability strategies. J Pharm Innov. 2014;9(4):259–71.
Freed AL, Clement E, Timpano R. Regulatory responses to the use of various lean stability strategies in early drug development. Regul Rapporteur. 2014;11(7):5–8.
ICH: Guideline Q1A(R2) (2003) Stability testing of new drug substances and products, Step 4 version. https://database.ich.org/sites/default/files/Q1A%28R2%29%20Step4.pdf
Scrivens G, Clancy D, Gerst P. Chapter 3 Theory and fundamentals of accelerated predictive stability (APS) studies. Accelerated Predictive Stability (APS): Fundamentals and Pharmaceutical Industry Practices; 2018. p. 33–73. https://doi.org/10.1016/C2014-0-02298-8.
Don C, Neil H, Rachel O, Martin O, John P. Kinetic model development for accelerated stability studies. AAPS PharmSciTech. 2017;18(4):1158–76.
Waterman KC. The application of the accelerated stability assessment program (ASAP) to quality by design (QbD) for drug product stability. AAPS PharmSciTech. 2011;12(3):932–7.
Li QC, Qiu F, Cohen K, Tougas T, Li J, McCaffrey J, Purdue T, Song JJ, Swanek F, Abelaira S (2012) Best practices for drug substance stress and stability studies during early-stage development part I – conducting drug substance solid stress to support phase 1a clinical trials. J Pharm Innov 7(3–4): 214–224.
Freed AL, Colgan ST, Kochling JD, Alasandro MS. AAPS Workshop: accelerating pharmaceutical development through predictive stability approaches, April 4–5, 2016. AAPS Open. 2017;3:8.
ICH: Guideline Q5C (1995) Stability testing of biotechnological/biological products, Step 4 version. https://database.ich.org/sites/default/files/Q5C_Guideline.pdf
ICH: Guideline Q1E (2003) Evaluation for stability data Q1E.” Current Step 4: February (2003). https://database.ich.org/sites/default/files/Q1E_Guideline.pdf
Funding
See Author’s Note above.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author’s Note
This paper was developed with the support of the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ, www.iqconsortium.org). IQ is a not-for-profit organization of pharmaceutical and biotechnology companies with a mission of advancing science and technology to augment the capability of member companies to develop transformational solutions that benefit patients, regulators and the broader research and development community.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adamec, E., Babayan, Y., Catacchio, B. et al. Lean Stability Case Studies—Leveraging Science- and Risk-Based Approaches to Enable Meaningful Phase Specific Pharmaceutical Stability Strategies. J Pharm Innov 16, 566–574 (2021). https://doi.org/10.1007/s12247-020-09463-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12247-020-09463-z