
Abstract
Hypoxia is common feature of eutrophic estuaries and semi-enclosed seas globally. One of the key factors driving hypoxia is nitrogen pollution. To gain more insight into the effects of hypoxia on estuarine nitrogen cycling, we measured potential nitrate reduction rates at different salinities and levels of hypoxia in a eutrophic temperate microtidal estuary, the Neuse River Estuary, North Carolina, USA. We also tested the effect of hydrogen sulfide and ferrous iron additions on the nitrate reduction pathways. Overall, DNRA dominated over denitrification in this periodically hypoxic estuary and there was no correlation between the potential nitrate reduction rates, salinity, or dissolved oxygen. However, when hypoxia lasted several months, denitrification capacity was almost completely lost, and nearly all nitrate added to the sediment was reduced via DNRA. Additions of hydrogen sulfide stimulated DNRA over denitrification. Additions of ferrous iron stimulated nitrate consumption; however, the end product of nitrate consumption was not clear. Interestingly, substantial nitrous oxide formation occurred in sediments that had experienced prolonged hypoxia and were amended with nitrate. Given expanding hypoxia predicted with climate change scenarios and the increasing nitrate loads to coastal systems, coastal sediments may lose their capability to mitigate nitrogen pollution due to DNRA dominating over denitrification during extended hypoxic periods.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The amount of reactive nitrogen (N) in the environment has increased dramatically during the past 150 years, which has promoted eutrophication of coastal waters (Paerl and Piehler 2008). One of the most severe symptoms of eutrophication is hypoxia (oxygen (O2) 90 μmol l−1 at STP) (Diaz and Rosenberg 1995, 2008; Breitburg et al. 2018), which alters biogeochemical cycling of numerous key elements (e.g., carbon, nitrogen, sulfur, phosphorus).
Microbes in coastal sediments provide an important ecosystem service by converting reactive N through microbial pathways to dinitrogen (N2) and nitrous oxide (N2O) gases (Seitzinger 1988; Dalsgaard et al. 2005). It has been estimated that globally, coastal and shelf areas remove approximately of 25% of the total fluvial reactive N input (Sharples et al. 2017). However, under hypoxic conditions, N removal in coastal sediments decreases, since nitrate (NO3−) is also reduced to ammonium (NH4+), instead of to N2 and N2O, via the dissimilatory nitrate reduction to ammonia (DNRA) pathway (An and Gardner 2002; Gardner et al. 2006; Dong et al. 2011). DNRA can be performed by fermentative heterotrophic organisms, which use organic carbon as the electron donor and by chemolithoautotrophic organisms, which use reduced inorganic compounds as an electron donor (Giblin et al. 2010). In general, fermentative processes are slow and fermentative DNRA cannot compete with dentification for NO3− unless the carbon the NO3− ratio is very high (Kraft et al. 2014). Chemolithoautotrophic DNRA in the other hand is not dependent on the availability of organic carbon (An and Gardner 2002; Gardner et al. 2006; Dong et al. 2011). Under benthic anoxia, sedimentary nitrification, which typically provides most substrate for the NO3− reducing processes (i.e., Hietanen and Kuparinen 2008), ceases and thus DNRA rates are also inhibited due to the low availability of electron acceptors. Hence, hypoxia can lead to a vicious cycle of eutrophication where the excess bioavailable N builds up, preventing recovery from this process (Jäntti and Hietanen 2012).
Sulfate (SO42−) is highly available in marine ecosystems (Canfield 1989 and references therein) and a common consequence of hypoxia is the accumulation of toxic hydrogen sulfide (H2S) in the bottom water. H2S is produced when, in the absence of O2, organic matter is oxidized with SO42− (Middelburg and Levin 2009). H2S affects sedimentary N cycling by inhibiting nitrification and anammox (Joye and Hollibaugh 1995; Hannig et al. 2007), while stimulating chemolithotrophic denitrification and DNRA (Brettar and Rheinheimer 1991; An and Gardner 2002; Gardner et al. 2006; Dong et al. 2011). Hence, hypoxia creates favorable conditions for DNRA; however, the presence of this process is still poorly quantified in estuarine systems.
H2S is often bound to iron (Fe) compounds, which can also be abundant in estuaries and coastal areas. The interactions between H2S, Fe, and various forms of N are currently poorly understood. The reactions between NO3−, nitrite (NO2−), and reduced Fe, which regulate the bioavailability of N, were initially considered to be mainly abiotic (Moraghan and Buresh 1977; Buresh and Moraghan 1976). However, Straub et al. (1996) showed that Fe oxidation with NO3− can also be microbially mediated and since then the number of microbial taxa known to be capable for anaerobic Fe2+ oxidation with NO3− under dark circumneutral conditions has increased substantially (Weber et al. 2006). The magnitude of microbial Fe oxidation with NO3− has, however, been questioned because of inappropriate analysis of N and Fe intermediates which can result in overestimated rates (Picardal 2012; Klueglein and Kappler 2013). Overall, the significance of Fe determining the relative importance of denitrification and DNRA in natural environments is currently poorly constrained.
The purpose of this study was to investigate the fate of NO3− in a eutrophic, seasonally hypoxic, N limited estuary. More specifically, we wanted to investigate how salinity and O2 concentration affect the NO3− reduction pathways and how elevated availabilities of reduced S and Fe species affect the interplay between the different NO3− reducing pathways under anoxic conditions.
Methods
Sampling
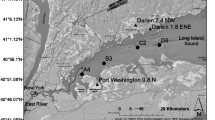
Samples for the sediment incubations were collected from the Neuse River Estuary (NRE). The NRE is the largest sub-estuary of the lagoonal Albemarle-Pamlico Sound, which is the second largest estuarine complex in the lower USA. The average depth of the NRE is ~ 3.5 m and the astronomical tidal range is < 0.1 m (Luettich Jr. et al. 2000). Sediment composition in the NRE changes from sandy at the shoreline to muddy in mid-channel. Its watershed drains rapidly expanding agricultural (animal and row crop operations), urban (Raleigh–Durham Research Triangle), and industrial centers within the piedmont and coastal plain regions of North Carolina. The NRE has over the past several decades undergone anthropogenic eutrophication leading to increased phytoplankton production and bloom frequency (Paerl et al. 2010). The saltwater wedge extending from the downstream Pamlico Sound up into the estuary naturally decreases ventilation of the benthic waters and resultant hypoxia is a seasonal (spring through fall) feature of this estuary (Paerl et al. 1998; Fear et al. 2005).
Intact sediment cores (diameter 20 cm, height 50 cm), utilized for determining potential NO3− reduction rates, were collected with a custom-made coring device from the muddy mid-channel from one station (St70) in May 2016, and two stations (St30 and St70) in October 2016 and June 2017 (Fig. 1). Surface sediment (0–1 cm) for the slurry incubations was collected in May 2016 from St70. Temperature and salinity were recorded 50 cm above the sediment surface, using a YSI 6800 multi-probe sonde (YSI incorporated, Yellow Springs, OH, USA). The O2, NO2− + NO3− (referred as NOx−) and NH4+ concentrations in the bottom water were determined by withdrawing a water sample from approximately 5 cm above the sediment surface in a core. The NOx− and NH4+ samples were filtered (prewashed 0.8/0.2-μm double filter syringe, Sarsted, Nümbrecht, Germany) and frozen at − 20 °C for subsequent flow-injection nutrient analysis (Lachat instruments, Loveland, CO, USA). Dissolved O2 concentration was measured using the Winkler method (Grasshoff et al. 1999).

Map of the Neuse Estuary in North Carolina, USA. The stations sampled in this study are marked as 30 and 70
Intact Core Incubations
For the potential NO3− reduction measurements, each large core was sub-sampled using three or four smaller acryl plastic cores (diameter 2.5 cm, height 18 cm), so that there was approximately 5 cm of sediment and 13 cm of water in each sub-sample leaving no headspace in the cores. The sediment samples from different casts were randomized for the treatments in which they were enriched with potassium nitrate (K15NO3−, 99% 15N, Sigma–Aldrich, St. Louis, MO, USA (2016), and K15NO3−, 99% 15N, Cambridge Isotope Laboratories, Andover, MA, USA (2017)) to final concentration of 0, 40, 80, 120, and 160 μM 15NO3− in the overlying water (n = 4 per 15NO3− concentration). Thereafter, the cores were pre-incubated for 15 min prior to closing the caps. The cores without the tracer were sacrificed immediately at beginning of the incubation to calculate background 15N2, 15N2O, and 15NH4+ concentrations. The capped cores were incubated in darkness for 3–4 h at in situ temperature with magnetic stirring bars placed in the caps. After incubation, the sediment was mixed with the overlying water and allowed to stand until most sediment particles had settled (typically 5–15 min). A 12-ml water sample for isotopic analysis of N2 and N2O was withdrawn from the top of the core into a gas tight glass vial (Exetainer, Labco Scientific, High Wycombe, UK) containing 0.5 ml ZnCl2 (100% w/v). The remaining core content was centrifuged (1500 rpm, 10 min) and the supernatant was filtered (prewashed 0.8/0.2-μm double filter syringe, Sarsted, Nümbrecht, Germany) and frozen at − 20 °C for subsequent and NH4+ and 15NH4+ analyses.
Slurry Incubations
To determine the effect of the availability of different electron donors on NO3− reduction rates, sediment-water slurries were prepared in May 2016 from sediment and water collected at St70. The slurry for the incubation experiments was comprised of the top 1 cm in the sediment cores and water collected directly above the sediment. The water and sediment were mixed at a 1:1 (vol:vol) ratio and purged with N2 for 15 min to remove O2 and background CO2 and H2S gases. Purging of the slurry was likely to increase the sample pH by 1–2 units due to CO2 leaving the sample. However, St70 experiences pH changes in that range when the saltwater wedge is intruding in the estuary; thus, the microbes present in the sample are likely to experience similar changes naturally. After purging, the slurry was transferred into a glove bag and handled under a N2 atmosphere until the incubation vials were capped. The slurry was divided into for four 1-l bottles for the following additions in the final concentration: (1) 1000 μM Na2S (H2S treatment); (2) 1000 μM Fe(II)SO4 (Fe2+ treatment); (3) 1000 μM Na2S + 1000 μM Fe(II)SO4 (H2S + Fe2+ treatment); (4) no additional electron donors (control treatment). All treatments received approximately 100 μM K15NO3−. The treatment solutions were prepared in N2 purged water and the Fe(II)SO4 addition was verified to reduce the pH of the slurries by less than 0.5. The slurry from each treatment bottle was divided into 32 12-ml gas tight glass vials (Exetainer, Labco Scientific, High Wycombe, UK), resulting in a total of 128 samples. After preparation, the samples were incubated for approximately 6 h. Then four samples from each treatment were terminated for 15N2 and 15N2O concentration measurements by creating a 4-ml He headspace and adding 100 μl of ZnCl2 (100% w/v) to each sample. Simultaneously, incubations of four samples from each treatment were terminated by filtering the sample through prewashed 0.8/0.2-μM double filter syringes (Acrodisc, Pall Scientific, New York, NY, USA). Thereafter, samples were terminated every 6 h, until 24 h of incubation. The filtrate was analyzed within few hours for NO3− and NO2−, (Fawcett and Scott 1960; Miranda et al. 2001) and the rest of the samples were frozen at − 20 °C and analyzed later for NH4+ and 15NH4+ concentrations.
15N Analyses
For the 15N2 and 15N2O gas samples, a 4 ml helium (He) headspace was created immediately before analysis and samples withdrawn from the headspace were analyzed with a ThermoScientific GasBench + Precon gas concentration system interfaced to a ThermoScientific Delta V Plus isotope-ratio mass spectrometer (ThermoScientific, Bremen, Germany) at the University of California Davis Stable Isotope Laboratory (Davis, CA, USA) (May 2016) and with Isoprime100 IRMS coupled to an Isoprime TraceGas pre-concentration unit (Elemental Analysensysteme GmbH, Langenselbold, Germany) at the University of Jyväskylä (Jyväskylä, Finland) (October 2016 and June 2017).
The 15NH4+ analysis was modified from Sigman et al. (1997) and Holmes et al. (1998). First, NH4+ concentrations of the samples were determined according Fawcett and Scott (1960) to ensure that a minimum of 10 μM NH4+ was available for the extraction. Then, 10 ml of sample was placed in a 20-ml HDPE scintillation vial (Wheaton, Millville, NJ, USA) and the salinity of the samples was adjusted to 30 with sodium chloride (NaCl). Thereafter, 100 mg of magnesium oxide (MgO) was added to each sample. pH was measured after the addition of MgO to ensure that it was optimal (~ 10) pH for the conversion of NH4+ to NH3. The liberated NH3 was collected in diffusion packets which were constructed by pipetting 30 μl of 2.5 M potassium bisulfate (KHSO4) on to a fiberglass filter (Whatman, GF/D, diameter 5 mm, Whatman, Maidstone, Kent, UK) that was placed between two pieces of Teflon tape. The diffusion packets were added prior to MgO addition to minimize NH3 escaping before closing the vials. The vials were incubated for 3 days at + 37 °C on a shaker table (150 rpm). Then, the diffusion packets were removed from the vials and placed in a desiccator under a sulfuric acid (H2SO4) atmosphere to dry. After 2 days, the packets were disassembled, and the fiberglass filters were packed into silver foil cups (Elemental Microanalyses Ltd., Toft, Cambridge, UK). The isotopic ratio of the extracted N was analyzed using Thermo Finnigan Delta V plus (Thermo Scientific, Waltham, MA, USA) at the University of Eastern Finland (Kuopio, Finland).
Calculations and Statistics
Our initial aim was to measure in situ NO3− reduction rates with the isotope pairing method (IPT; Nielsen 1992, Christensen et al. 2000, Risgaard-Petersen et al. 2003, 2004, Master et al. 2005). However, two problems arose during the measurements: (1) The concentration of 15N-labelled N species did not always linearly increase with the 15NO3− concentration (supplementary figure) and (2) the O2 concentration in the cores during all sampling times was so low that although the incubation time was kept as short as possible, the cores were nearly anoxic by the end of the incubation. These artifacts violate the fundamental assumptions behind the IPT (Nielsen 1992), and calculation of reliable in situ NO3− reduction rates was impossible. Therefore, we calculated only the potential NO3− reduction rates be averaging the 15N production rates from all 15NO3− concentrations.
Potential N2 production is the average of excess 15N2 (p29N2 + 2 × 30N2), potential N2O production is the average of excess 15N2O (p45N2O + 2 × p46N2O), and potential DNRA is the average excess 15NH4+ production. Because in June 2017 there was an increasing trend in the 15N2 production with the 15NO3− concentration, the potential during that sampling time provides only a minimum estimate and the actual potentials might be higher (supplementary figure). The differences in rates measured in the core samples were tested by using Whitney–Mann U test. The correlations between the potential rates and environmental parameters were determined by using non-parametric correlation analysis.
The NO3− consumption and NO2−, 15N2, 15N2O, and 15NH4+ production rates were calculated from liner regression analysis between the N concentration of different N-species and time. Rates were considered significant at p = 0.05. The comparison between the rates was done by using two-way ANOVA.
Results
Environmental Conditions
Both stations were hypoxic during all sampling times. However, there was always a small amount O2 detected, except in Oct 2016 at st70 when the odor of H2S was detected in the bottom water and in May 2017 at St30 when O2 was at the detection limit (~ 3 μmol l−1) of the Winkler method (Table 1).
In May 2016, the bottom water salinity was at its lowest at St70, most likely due to excessive rainfall and elevated freshwater runoff which pushed the salt wedge towards the outlet of the estuary. In October 2016, the bottom water salinity at St70 was higher compared with May; however, the saltwater wedge did not reach deep into the estuary and the salinity at St30 was 0. In June 2017, when the river flow had remained low and the saltwater wedge intruded deep into the estuary, the bottom water salinity was 12 at St70 and 10 at St30 (Table 1). The bottom water NOx− concentration was always higher at St30 than at St70, but the opposite was true for NH4+ (Table 1).
Potential Denitrification and DNRA Rates Measured on the Intact Cores
There was no evidence of anammox based on the IPT calculations (Risgaard-Petersen et al. 2003, 2004); hence, most N2 must have originated from denitrification. Occasionally 1–2 samples were lost during sample processing, and consequently there was 14–16 samples analyzed per station per sampling time. During all sampling times, the potential DNRA rates were equal (June 2017, St30, n = 16, p = 0.861) or significantly higher (May 2016, St70, n = 15, p = 0.050; October 2016, St70, n = 15, p = 0.000; October 2016, St30, n = 14, p = 0.000; June 2017, St70, n = 16, p = 0.008) than the potential denitrification (N2O + N2) rates at both stations. The total NO3− reduction rates (denitrification + DNRA) were always significantly lower at St30 than at St70 (October 2016, n = 15, p = 0.000; June 2017, n = 16, p = 0.000) (Fig. 2). In October 2016, potential N2 production was detected in only in seven samples at St70 and at twelve samples at St70, while the rest were below the detection limit. 15N2O production was detected in 12 samples St70 and at all samples at St30. Where denitrification was detected, over 50% of it was N2O (Table 1). However, potential DNRA was still proceeding at measurable rates at both stations. There was no significant difference in denitrification and DNRA rates between May 2016 and June 2017 (n = 15 in May 2016 St70 and n = 16 in June 2017 St70, p = 0.545 for denitrification and p = 0.866 for DNRA) (Fig. 2). There were no statistically significant correlations (p < 0.05) between the production of 15N-labelled products and environmental parameters (O2, temperature and salinity) in the core samples.

The denitrification (15N2 and 15N2O) and DNRA (15NH4+) potentials measured from the intact sediment cores
Potential Denitrification and DNRA Rates Measured from Slurry Samples
The addition of 1 mM H2S increased the 15NO3− consumption rates, although not significantly, when compared with the control samples and with samples that had H2S + Fe2+ (Table 2). Although the total 15N2 concentration remained lower than in the control samples, it was still increasing steadily over the entire incubation period (Fig. 3). There was also a steady increase in the 15NH4+ concentration and the total 15NH4+ production rates were significantly higher in the samples that were amended with H2S compared with all other treatments (Table 2). However, H2S did not inhibit denitrification completely (Table 2, Fig. 3).

Concentrations of 15N2, 15N2O, 15NH4+, NO2−, and NO3− over the incubation period from slurry samples that were amended with different electron donors. The control samples had no added electron donors. All samples initially contained approximately 100 μM 15NO3−
Amendment of Fe2+ stimulated the NO3− consumption a nearly sixfold compared with the control samples (Table 2, Fig. 3). However, the sink of NO3− is unclear because after the initial increases in the concentrations of 15N2 and 15NH4+, their concentrations remained steady (15N2) or increased only moderately (15N2O, 15NH4+) over the entire incubation period, although NO3− consumption continued at a high rate (Table 2, Fig. 3).
The samples which were amended with both H2S and Fe2+ had slightly lower 15N2 production rates and slightly higher NO3− consumption rates when compared with the control samples. This and the high NO3− consumption in the Fe2+-amended samples indicate that Fe2+ stimulates NO3− consumption regardless whether H2S is present or not (Table 2, Fig. 3).
The NO2− concentrations remained low in samples that were treated with either H2S or Fe2+. However, when H2S and Fe2+ were added in equal concentrations, NO2− concentration was higher than in the samples treated with only H2S or Fe2+ but lower than in the control samples. Interestingly, concentrations and production rates of N2O followed NO2− concentrations and production rates and they all decreased at the last sampling point, except in the H2S treatments where the NO3− concentration was approximately double as high compared with other treatments (Table 2, Fig. 3). The 15NH4+ product increased, although not significantly, simultaneously to the decrease in NO2− concentration at the last sampling point in the control samples and H2S + Fe2+-treated samples (Fig. 3).
Discussion
NO3 − Reduction Rates in the Intact Cores
Overall, potential denitrification rates measured in the NRE were on the lower end of the range of average in situ denitrification rates in temperate coastal sediments (1.2–5 mmol m2 day−1; Seitzinger 1988) but within the same range (0–6600 μM N m−2 day−1) previously measured in the NRE by Fear et al. (2005). The low rates in this study can be explained by sampling during warm months when hypoxia is present, which is when Fear et al. (2005) also measured the lowest denitrification rates.
Although denitrification is an anoxic process, there typically is a positive correlation between denitrification rates and O2 concentration because nitrification, which provides NO3− for denitrification, is stimulated by the availability of O2 (Jenkins and Kemp 1984; Kemp et al. 1990). Since we only measured potentials, where denitrification is not limited by the availability of NO3−, we expected to find a negative correlation between denitrification rates and O2 concentration. However, no such correlation was found, and this may be because under hypoxia the presence of H2S, rather than the negligible availability of O2, is likely to be the key regulating factor for denitrification.
N2O production in core samples collected in October 2016 was on the higher end of measured sedimentary N2O fluxes in estuarine environments (Murray et al. 2015 and the references there in). This can be partially explained by it being a potential rather than in situ rate since there was more NO3− available than under naturally occurring conditions. Presence of H2S in the bottom water clearly favored N2O production over N2 production, since 99% of end product of denitrification was N2O (Table 1). This is consistent with the early findings of Sørensen et al. (1980) reporting inhibition of N2O reduction causing accumulation of N2O in the presence of H2S.
The potential DNRA rates measured at St70 are comparable with DNRA rates measured in the other estuaries in the southern USA (0–2.4 mmol N m−2 day−1; Gardner et al. 2006); (0–8.2 mmol N m−2 day−1; Giblin et al. 2010) as well as rates measured in sulfide-rich sediments in Denmark (0–6.5 mmol N day−1, Christensen et al. 2000). At St30, potential DNRA rates were substantially lower but still comparable with DNRA rates measured in periodically anoxic Baltic Sea sediments (0.03–1.1 mmol N m−2 day−1; Jäntti and Hietanen 2012). Surprisingly, there was also no correlation between the DNRA and O2 concentration, and the highest DNRA rates were measured under conditions when little O2 was available (May 2016). This may be because NO3− reduction to NO2−, the first step of DNRA, is suggested to be also be driven by denitrifying bacteria (Canfield et al. 2005) which can be inhibited by H2S. Hence, the DNRA rates were probably limited by the NO2− availability. It appears that if H2S is present near denitrification layer, the overall NO3− reduction rates are lower than in conditions when H2S is not present. However, the lowest denitrification to DNRA ratio was found when H2S was present in the bottom water (Oct 2016) suggesting that the presence of H2S inhibited denitrification relatively more than DNRA.
Salinity did not correlate with any of the 15N production rates, although increases in salinity have been linked to stimulation of DNRA (Gardner et al. 2006; Giblin et al. 2010). Consequently, it appears that in the NRE, the duration of hypoxia, rather than salinity or O2 availability, determines the dominance of DNRA over denitrification.
NO3 − Reduction Rates in the Sediment Slurry Samples
In the slurry samples, the slopes of N2 production and NO3− consumption proceeded steadily but in opposite directions in the control and H2S + Fe2+ treatments, indicating presence of denitrification. Similar 15N2 production and NO3− consumption pattern between the control and H2S + Fe2+ treatments can be explained by H2S chemically reacting with Fe2+ and forming iron sulfide (FeS), which is a relatively stable compound and does not act as an electron donor for either DNRA or denitrification (Brunet and Garcia-Gil 1996). However, the 15NH4+ concentration was higher in the H2S + Fe2+-treated samples compared with the control samples and it appears that presence of additional electron donors caused a higher percentage of NO3− to be converted to NH4+ over the entire incubation period. Because there was no NO2− accumulation and substantial 15NH4+ accumulation in the H2S + Fe2+-treated samples, it is likely that the additional electron acceptors stimulated NO2− reduction, the end product of which is NH4+. The simultaneously elevated concentrations of NO2− and N2O in nearly all treatments are in line with the results from wastewater treatment facilities where high N2O production during the denitrification phase has been linked with elevated NO2− concentrations, although the explanation for this relationship is not clear (Kampschreur et al. 2009).While NO2− and N2O concentrations began to decrease during the last measurement time, the 15NH4+ concentration began to increase, although the increase was not linearly significant, suggesting that DNRA has a higher affinity for NO2− than denitrification, similar to results of Kraft et al. (2014). However, the opposite has been observed in chemostat experiments (van den Berg et al. 2017), suggesting that the dominant NO2− reduction pathway is also dependent on NO2− reducing microbial community composition.
In the H2S-treated slurries, there was a steady increase in 15N2, suggesting that denitrifying microbes were still reducing NO3−, although at rates lower than the controls and almost equal proportions of NO3− were reduced to N2 and NH4+. This was expected, since stimulation of DNRA under elevated H2S concentrations has been previously demonstrated in several studies (Høgslund et al. 2009; Schutte et al. 2018; An and Gardner 2002; McCarthy et al. 2008; Jäntti and Hietanen 2012; Bernard et al. 2015; Murphy et al. 2020), as well as in our core incubations, where the highest DNRA to denitrification ratio was observed when the bottom water contained H2S in October 2016. However, the NO3− concentrations remained higher in the H2S-treated samples than in the control and H2S + Fe2+-treated samples, suggesting that H2S overall inhibits NO3− reduction, particularly immediately after it becomes present. This was indicated by much lower decrease in the NO3− concentration at the first 6 h of incubation in the H2S-treated samples when compared with the control samples. Interestingly, the N2O accumulation in the H2S-treated samples was not substantially higher than in the control samples, although N2O accumulation was observed in the intact cores in presence of H2S. This could be explained by slurries being in a closed incubation system where intermediates, such as N2O, cannot escape from the active site (sediment surface) during the incubation, unlike in the intact cores and consequently the intermediates in denitrification/DNRA were consumed at the rate which they were formed.
The effect of Fe2+ on N cycling is not yet well understood because it is challenging to separate the roles of abiotic and biotic processes since they can combine, even within a single organism (Picardal 2012; Melton et al. 2014; Ionescu et al. 2015). Also, most studies have been done by using microbial cultures, and the role of micro-organisms oxidizing Fe2+ with NO3− in natural environments is currently poorly known. In the slurry samples, there was a rapid accumulation of 15N2 at beginning of the incubation in Fe2+-treated samples. However, the concentration of 15N2 did not change after the initial increase; hence, denitrification was quickly brought to a halt. The rapid decrease in N2 formation in the Fe2+-treated samples could be caused by Fe2+ reacting with the organic carbon compounds present in the sediment producing poorly degradable compounds (Lalonde et al. 2012; Shields et al. 2016), and thus the low availability of labile organic carbon limited heterotrophic denitrification shortly after Fe2+ was introduced. Fe2+ can also disturb intracellular electron transport (Carlson et al. 2012) that inhibits denitrification rates and could explain the non-linear increase of 15N2 in the Fe2+-treated slurries.
The low 15NH4+ formation in the Fe2+-treated samples was unexpected as several studies have demonstrated that Fe2+ stimulates microbe-mediated DNRA (Robertson et al. 2016; Robertson and Thamdrup 2017; Kessler et al. 2018; Kessler et al. 2019) and abiotic NO3− reduction to NH4+ (Hansen et al. 1994; Hansen et al. 1996; Guerbois et al. 2014). Hence, DNRA in sediments of at St70 appears to be driven by H2S rather than Fe2+. This could be because the hypoxia is severe and long-lasting in the NRE (Buzzelli et al. 2002), causing H2S to rise regularly to the sediment surface and thus favoring microbes that can tolerate and utilize H2S in their metabolism. The reports of Fe2+ stimulating DNRA are mainly from sediments where free H2S is not available due to Fe2+ binding the free sulfides (Robertson et al. 2016; Robertson and Thamdrup 2017; Kessler et al. 2018). Also, there was no visible sign of the presence of cable bacteria which have been shown to promote DNRA through Fe-sulfide dissolution (Kessler et al. 2019).
There was the steep negative slope of NO3− concentration in the Fe2+-treated slurries and the NO3–N was not recovered from the N2, N2O, or from the DIN pools. One possible explanation for the disappearance of NO3− could be formation of nitric oxide (NO), which has been reported in the presence of Fe2+ (Carlson et al. 2012 and references therein). Since NO is highly volatile, it can be released immediately after opening the incubation vials. Also, because NO is highly reactive with metal oxides and formation of metal nitrosyls during the incubation, this cannot be excluded. It appears that additional chemical reactions between NO3− and Fe2+ can play a significant role in the sedimentary coastal N cycling and deserve further investigations.
Lack of NO2− accumulation in the Fe2+- and H2S-treated samples indicates that the additional electron acceptors particularly stimulate NO2− reduction rates. In the H2S-treated samples, NO2− was likely to be reduced to NH4+, whereas in the Fe2+-treated samples, the end product of NO2− reduction is not clear. Both NO2− and Fe2+ are highly reactive, and one of the best documented reactions between NO2− and Fe2+ is the formation of N2O (Moraghan and Buresh 1977). However, neither N2O production nor changes in concentrations of the other measured N species match the NO3− consumption rates. Hence, unmeasured processes, such as NO formation, seem more likely.
Conclusions
Based on the results of this and other experiments, we conclude that the length of hypoxia has a substantial effect on the N cycling processes in carbon and SO42− rich sediments and that the role of Fe2+ has to investigated more thoroughly in hypoxic estuarine sediments where H2S accumulation occurs. When O2 is present in the sediment surface (Fig. 4A), nitrification proceeds at the rate which NH4+ is produced by mineralization. There is no NH4+ release in the bottom water because nitrification efficiently oxidizes the produced NH4+, and denitrification is tightly coupled to nitrification. The DNRA rates are low because in the absence of H2S, aerobic heterotrophy dominates, and DNRA microbes cannot compete with denitrification because the quality of organic carbon available for anaerobic processes, after intensive aerobic processes, is low (Kraft et al. 2014). When hypoxia first settles in (Fig. 4B), nitrification proceeds at high rates because nitrification can tolerate microaerophilic conditions (Laanbroek and Gerards 1993; Jäntti et al. 2018) although the end product of nitrification can switch to N2O (Kalvelage et al. 2011). The denitrification rates increase because denitrification is stimulated by decreasing O2 concentrations (Hietanen and Lukkari 2007). At this stage, availability of H2S near the sediment surface is still low, and consequently chemolithotrophic DNRA rates remain moderate. When hypoxia is established (Fig. 4C), H2S reaches sediment surface and nitrification ceases because it does not tolerate H2S (Joye and Hollibaugh 1995). Denitrification rates decrease and the end product of denitrification changes from N2 to N2O. At the same time, DNRA begins to dominate NO3− reduction. During persistent hypoxia (Fig. 4D), H2S reaches bottom water, and there is no nitrification and only very little denitrification. DNRA rates also decrease because of low NO2− availability.

The effect of length of hypoxia on NO3− reduction in eutrophic SO42−-rich estuarine sediments
References
An, S., and W.S. Gardner. 2002. Dissimilatory nitrate reduction to ammonium (DNRA) as a nitrogen link, versus denitrification as a sink in a shallow estuary (Laguna Madre/Baffin Bay, Texas). Marine Ecology Progress Series 237: 41–50. https://doi.org/10.3354/meps237041.
Bernard, R.J., B. Mortazavi, and A.A. Kleinhuizen. 2015. Dissimilatory nitrate reduction to ammonium (DNRA) seasonally dominates NO3− reduction pathways in an anthropogenically impacted sub-tropical coastal lagoon. Biogeochemistry 125 (1): 47–64. https://doi.org/10.1007/s10533-015-0111-6.
Breitburg, D., L.A. Levin, A. Oschlies, M. Grégoire, F.P. Chavez, D.J. Conley, V. Garçon, D. Gilbert, D. Gutiérrez, K. Isensee, G.S. Jacinto, K.E. Limburg, I. Montes, S.W.A. Naqvi, G.C. Pitcher, N.N. Rabalais, M.R. Roman, K.A. Rose, B.A. Seibel, M. Telszewski, M. Yasuhara, and J. Zhang. 2018. Declining oxygen in the global ocean and coastal waters. Science 359 (6371): eaam7240. https://doi.org/10.1126/science.aam7240.
Brettar, I., and G. Rheinheimer. 1991. Denitrification in the Central Baltic: evidence for H2S-oxidation as motor of denitrification at the oxic-anoxic interface. Marine Ecology Progress Series 77: 157–169.
Brunet, R.C., and L.J. Garcia-Gil. 1996. Sulfide-induced dissimilatory nitrate reduction to ammonia in anaerobic freshwater sediments. FEMS Microbiology Ecology 21 (2): 131–138. https://doi.org/10.1111/j.1574-6941.1996.tb00340.x.
Buresh, R.J., and J.T. Moraghan. 1976. Chemical reduction of nitrate by ferrous iron 1. Journal of Environmental Quality 5 (3): 320–325. https://doi.org/10.2134/jeq1976.00472425000500030021x.
Buzzelli, C.P., S.P. Powers, R.A. Luettich Jr., J.E. McNinch, C.H. Peterson, J.L. Pinckney, and H.W. Paerl. 2002. Estimating the spatial extent of bottom water hypoxia and benthic fishery habitat degradation in the Neuse River Estuary, NC. Mar Ecol Progr Ser 230: 103–112.
Canfield, D.E. 1989. Sulfate reduction and oxic respiration in marine sediments: implications for organic carbon preservation in euxinic environments. Deep Sea Res A 36 (1): 121–138.
Canfield, D.E., E. Kristensen, and B. Thamdrup. 2005. Aquatic geomicrobiology. New York: Academic Press.
Carlson, H.K., I.C. Clark, R.A. Melnyk, and J.D. Coates. 2012. Toward a mechanistic understanding of anaerobic nitrate-dependent iron oxidation: balancing electron uptake and detoxification. Frontiers in Microbiology 3: 57. https://doi.org/10.3389/fmicb.2012.00057.
Christensen, P.B., S. Rysgaard, N.P. Sloth, T. Dalsgaard, and S. Schwærter. 2000. Sediment mineralization, nutrient fluxes, denitrification and dissimilatory nitrate reduction to ammonium in an estuarine fjord with sea cage trout farms. Aquatic Microbial Ecology 21: 73–84. https://doi.org/10.3354/ame021073.
Dalsgaard, T., B. Thamdrup, and D.E. Canfield. 2005. Anaerobic ammonium oxidation (anammox) in the marine environment. Research in Microbiology 156 (4): 457–464. https://doi.org/10.1016/j.resmic.2005.01.011.
Diaz, R.J., and R. Rosenberg. 1995. Marine benthic hypoxia: a review of its ecological effects and the behavioural responses of benthic macrofauna. Oceanography and Marine Biology 33: 245–303.
Diaz, R.J., and R. Rosenberg. 2008. Spreading dead zones and consequences for marine ecosystems. Science 321 (5891): 926–929. https://doi.org/10.1126/science.1156401.
Dong, L.F., M.N. Sobey, C.J. Smith, I. Rusmana, W. Phillips, A. Stott, A.M. Osborn, and D.B. Nedwell. 2011. Dissimilatory reduction of nitrate to ammonium, not denitrification or anammox, dominates benthic nitrate reduction in tropical estuaries. Limnology and Oceanography 56 (1): 279–291. https://doi.org/10.4319/lo.2011.56.1.0279.
Fawcett, J.K., and J. Scott. 1960. A rapid and precise method for the determination of urea. Journal of Clinical Pathology 13 (2): 156–159.
Fear, J.M., S.P. Thompson, T.E. Gallo, and H.W. Paerl. 2005. Denitrification rates measured along a salinity gradient in the eutrophic Neuse River Estuary, North Carolina, USA. Estuaries 28 (4): 608–619. https://doi.org/10.1007/BF02696071.
Gardner, W.S., M.J. McCarthy, S. An, D. Sobolev, K.S. Sell, and D. Brock. 2006. Nitrogen fixation and dissimilatory nitrate reduction to ammonium (DNRA) support nitrogen dynamics in Texas estuaries. Limnology and Oceanography 51 (1part2): 558–568. https://doi.org/10.4319/lo.2006.51.1_part_2.0558.
Giblin, A.E., N.B. Weston, G.T. Banta, J. Tucker, and C.S. Hopkinson. 2010. The effects of salinity on nitrogen losses from an oligohaline estuarine sediment. Estuaries and Coasts 33 (5): 1054–1068. https://doi.org/10.1007/s12237-010-9280-7.
Grasshoff, K., K. Kremling, and M. Ehrhardt. 1999. Methods of seawater analysis. 3rd ed. Weinheim: Verlag Chemie GmbH.
Guerbois, D., G. Ona-Nguema, G. Morin, M. Abdelmoula, A.M. Laverman, J.M. Mouchel, K. Barthelemy, F. Maillot, and J. Brest. 2014. Nitrite reduction by biogenic hydroxycarbonate green rusts: evidence for hydroxy-nitrite green rust formation as an intermediate reaction product. Environmental Science & Technology 48 (8): 4505–4514. https://doi.org/10.1021/es404009k.
Hannig, M., G. Lavik, M.M.M. Kuypers, D. Woebken, W. Martens-Habbena, and K. Jürgens. 2007. Shift from denitrification to anammox after inflow events in the Central Baltic Sea. Limnology and Oceanography 52 (4): 1336–1345. https://doi.org/10.4319/lo.2007.52.4.1336.
Hansen, H.C.B., O.K. Borggaard, and J. Sørensen. 1994. Evaluation of the free energy of formation of Fe (II)-Fe (III) hydroxide-sulphate (green rust) and its reduction of nitrite. Geochimica et Cosmochimica Acta 58 (12): 2599–2608. https://doi.org/10.1016/0016-7037(94)90131-7.
Hansen, H.C.B., C.B. Koch, H. Nancke-Krogh, O.K. Borggaard, and J. Sørensen. 1996. Abiotic nitrate reduction to ammonium: key role of green rust. Environmental Science & Technology 30 (6): 2053–2056. https://doi.org/10.1021/es950844w.
Hietanen, S., and J. Kuparinen. 2008. Seasonal and short-term variation in denitrification and anammox at a coastal station on the Gulf of Finland, Baltic Sea. Hydrobiologia 596 (1): 67–77.
Hietanen, S., and K. Lukkari. 2007. Effects of short-term anoxia on benthic denitrification, nutrient fluxes and phosphorus forms in coastal Baltic sediment. Aquatic Microbial Ecology 49: 293–302. https://doi.org/10.3354/ame01146.
Høgslund, S., N.P. Revsbech, J.G. Kuenen, B.B. Jørgensen, V.A. Gallardo, J. Van De Vossenberg, J. Lund Nielsen, L. Holmkvist, E.T. Arning, and L.P. Nielsen. 2009. Physiology and behaviour of marine Thioploca. The ISME Journal 3 (6): 647–657. https://doi.org/10.1038/ismej.2009.17.
Holmes, R.M., J.W. McClelland, D.M. Sigman, B. Fry, and B.J. Peterson. 1998. Measuring 15N–NH4+ in marine, estuarine and fresh waters: an adaptation of the ammonia diffusion method for samples with low ammonium concentrations. Marine Chemistry 60 (3-4): 235–243. https://doi.org/10.1016/S0304-4203(97)00099-6.
Ionescu, D., C. Heim, L. Polerecky, V. Thiel, and D. De Beer. 2015. Biotic and abiotic oxidation and reduction of iron at circumneutral pH are inseparable processes under natural conditions. Geomicrobiology Journal 32 (3-4): 221–230. https://doi.org/10.1080/01490451.2014.887393.
Jäntti, H., and S. Hietanen. 2012. The effects of hypoxia on sediment nitrogen cycling in the Baltic Sea. AMBIO 41 (2): 161–169. https://doi.org/10.1007/s13280-011-0233-6.
Jäntti, H., B.B. Ward, J.W. Dippner, and S. Hietanen. 2018. Nitrification and the ammonia-oxidizing communities in the Central Baltic Sea water column. Estuarine, Coastal and Shelf Science 202: 280–289. https://doi.org/10.1016/j.ecss.2018.01.019.
Jenkins, M.C., and W.M. Kemp. 1984. The coupling of nitrification and denitrification in two estuarine sediments. Limnology and Oceanography 29 (3): 609–619. https://doi.org/10.4319/lo.1984.29.3.0609.
Joye, S.B., and J.T. Hollibaugh. 1995. Influence of sulfide inhibition of nitrification on nitrogen regeneration in sediments. Science 270 (5236): 623–625.
Kalvelage, T., M.M. Jensen, S. Contreras, N.P. Revsbech, P. Lam, M. Günter, J. LaRoche, G. Lavik, and M.M.M. Kuypers. 2011. Oxygen sensitivity of anammox and coupled N-cycle processes in oxygen minimum zones. PLoS One 6 (12): e29299. https://doi.org/10.1371/journal.pone.0029299.
Kampschreur, M.J., H. Temmink, R. Kleerebezem, M.S. Jetten, and M.C. van Loosdrecht. 2009. Nitrous oxide emission during wastewater treatment. Water Research 43 (17): 4093–4103. https://doi.org/10.1016/j.watres.2009.03.001.
Kemp, W.M., P. Sampou, J. Caffrey, M. Mayer, K. Henriksen, and W.R. Boynton. 1990. Ammonium recycling versus denitrification in Chesapeake Bay sediments. Limnology and Oceanography 35 (7): 1545–1563. https://doi.org/10.4319/lo.1990.35.7.1545.
Kessler, A.J., K.L. Roberts, A. Bissett, and P.L. Cook. 2018. Biogeochemical controls on the relative importance of denitrification and dissimilatory nitrate reduction to ammonium in estuaries. Global Biogeochem Cycles 32 (7): 1045–1057. https://doi.org/10.1029/2018GB005908.
Kessler, A.J., M. Wawryk, U. Marzocchi, K.L. Roberts, W.W. Wong, N. Risgaard-Petersen, F.J.R. Meysman, R.N. Glud, and P.L. Cook. 2019. Cable bacteria promote DNRA through iron sulfide dissolution. Limnology and Oceanography 64 (3): 1228–1238. https://doi.org/10.1002/lno.11110.
Klueglein, N., and A. Kappler. 2013. Abiotic oxidation of Fe (II) by reactive nitrogen species in cultures of the nitrate-reducing Fe (II) oxidizer A cidovorax sp. BoFeN1–questioning the existence of enzymatic Fe (II) oxidation. Geobiology 11 (2): 180–190. https://doi.org/10.1111/gbi.12019.
Kraft, B., H.E. Tegetmeyer, R. Sharma, M.G. Klotz, T.G. Ferdelman, R.L. Hettich, J.S. Geelhoed, and M. Strous. 2014. The environmental controls that govern the end product of bacterial nitrate respiration. Science 345 (6197): 676–679. https://doi.org/10.1126/science.1254070.
Laanbroek, H.J., and S. Gerards. 1993. Competition for limiting amounts of oxygen between Nitrosomonas europaea and Nitrobacter winogradskyi grown in mixed continuous cultures. Archives of Microbiology 159 (5): 453–459.
Lalonde, K., A. Mucci, A. Ouellet, and Y. Gélinas. 2012. Preservation of organic matter in sediments promoted by iron. Nature 483 (7388): 198–200. https://doi.org/10.1038/nature10855.
Luettich, R.A. Jr., J.E. McNinch, H.W. Paerl, C.H. Peterson, J.T. Wells, M.J. Alperin, C. S. Martens, and J.L. Pinckney. 2000. Neuse River Estuary modeling and monitoring project stage 1: hydrography and circulation, water column nutrients and productivity, sedimentary processes and benthic-pelagic coupling. Report UNC-WRRI-2000-325B, Water Resources Research Institute of the University of North Carolina, Raleigh, NC, 172p.
McCarthy, M.J., K.S. McNeal, J.W. Morse, and W.S. Gardner. 2008. Bottom-water hypoxia effects on sediment–water interface nitrogen transformations in a seasonally hypoxic, shallow bay (Corpus Christi Bay, TX, USA). Estuaries and Coasts 31: 521–531. https://doi.org/10.1007/s12237-008-9041-z.
Master, Y., U. Shavit, and A. Shaviv. 2005. Modified isotope pairing technique to study N transformations in polluted aquatic systems: Theory. Environmental Science & Technology 39 (6): 1749–1756. https://doi.org/10.1021/es049086c.
Melton, E.D., E.D. Swanner, S. Behrens, C. Schmidt, and A. Kappler. 2014. The interplay of microbially mediated and abiotic reactions in the biogeochemical Fe cycle. Nature Reviews Microbiology 12 (10): 797–3303. https://doi.org/10.1111/1462-2920.12566.
Middelburg, J.J., and L.A. Levin. 2009. Coastal hypoxia and sediment biogeochemistry. Biogeosciences 6 (7): 1273–1293. https://doi.org/10.5194/bg-6-1273-2009.
Miranda, K.M., M.G. Espey, and D.A. Wink. 2001. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 5 (1): 62–71. https://doi.org/10.1006/niox.2000.0319.
Moraghan, J.T., and R.J. Buresh. 1977. Chemical reduction of nitrite and nitrous oxide by ferrous iron 1. Journal of Environmental Quality 41 (1): 47–50. https://doi.org/10.2136/sssaj1977.03615995004100010017x.
Murphy, A.E., A.N. Bulseco, R. Ackerman, J.H. Vineis, and J.L. Bowen. 2020. Sulphide addition favours respiratory ammonification (DNRA) over complete denitrification and alters the active microbial community in salt marsh sediments. Environmental Microbiology (in press) 22 (6): 2124–2139. https://doi.org/10.1111/1462-2920.14969.
Murray, R.H., D.V. Erler, and B.D. Eyre. 2015. Nitrous oxide fluxes in estuarine environments: response to global change. Glob Change Biol 21 (9): 32193245–32193245. https://doi.org/10.1111/gcb.12923.
Nielsen, L.P. 1992. Denitrification in sediment determined from nitrogen isotope pairing. FEMS Microbiology Letters 86 (4): 357–361. https://doi.org/10.1111/j.1574-6941.1992.tb01771.x.
Paerl, H.W., J.L. Pinckney, J.M. Fear, and B.L. Peierls. 1998. Ecosystem responses to internal and watershed organic matter loading: consequences for hypoxia in the eutrophying Neuse River Estuary, North Carolina, USA. Marine Ecology Progress Series 166: 17–25. https://doi.org/10.3354/meps166017.
Paerl, H.W., and M.F. Piehler. 2008. Nitrogen and marine eutrophication. In Nitrogen in the marine environment, ed. D.G. Capone, M. Mulholland, and E. Carpenter, vol. 2, 529–567. Orlando: Academic Press.
Paerl, H.W., K.L. Rossignol, S.N. Hall, B.L. Peierls, and M.S. Wetz. 2010. Phytoplankton community indicators of short-and long-term ecological change in the anthropogenically and climatically impacted Neuse River Estuary, North Carolina, USA. Estuaries and Coasts 33 (2): 485–497. https://doi.org/10.1007/s12237-009-9137-0.
Picardal, F. 2012. Abiotic and microbial interactions during anaerobic transformations of Fe (II) and NOx. Frontiers in Microbiology 3: 112. https://doi.org/10.3389/fmicb.2012.00112.
Risgaard-Petersen, N., L.P. Nielsen, S. Rysgaard, T. Dalsgaard, and R.L. Meyer. 2003. Application of the isotope pairing technique in sediments where anammox and denitrification coexist. Limnology and Oceanography: Methods 1 (1): 63–73. https://doi.org/10.4319/lom.2003.1.63.
Risgaard-Petersen, N., L.P. Nielsen, S. Rysgaard, T. Dalsgaard, and R.L. Meyer. 2004. Erratum: application of the isotope pairing technique in sediments where anammox and denitrification coexist. Limnology and Oceanography: Methods 2 (9): 315. https://doi.org/10.4319/lom.2004.2.315.
Robertson, E.K., K.L. Roberts, L.D. Burdorf, P. Cook, B. and Thamdrup. 2016. Dissimilatory nitrate reduction to ammonium coupled to Fe (II) oxidation in sediments of a periodically hypoxic estuary. Limnology and Oceanography 61: 365–381. https://doi.org/10.1002/lno.10220, 1.
Robertson, E.K., and B. Thamdrup. 2017. The fate of nitrogen is linked to iron (II) availability in a freshwater lake sediment. Geochimica et Cosmochimica Acta 205: 84–99. https://doi.org/10.1016/j.gca.2017.02.014.
Schutte, C.A., A. Teske, B.J. MacGregor, V. Salman-Carvalho, G. Lavik, P. Hach, and D. de Beer. 2018. Filamentous giant Beggiatoaceae from Guaymas Basin are capable of both denitrification and dissimilatory nitrate reduction to ammonium (DNRA). Applied and Environmental Microbiology 84 (15): e02860–e02817. https://doi.org/10.1128/AEM.02860-17.
Seitzinger, S.P. 1988. Denitrification in freshwater and coastal marine ecosystems: ecological and geochemical significance. Limnology and Oceanography 33 (4part2): 702–724. https://doi.org/10.4319/lo.1988.33.4part2.0702.
Sharples, J., J.J. Middelburg, K. Fennel, and T.D. Jickells. 2017. What proportion of riverine nutrients reaches the open ocean? Global Biogeochem Cycles 31 (1): 39–58. https://doi.org/10.1002/2016GB005483.
Shields, M.R., T.S. Bianchi, Y. Gelinas, M.A. Allison, and R.R. Twilley. 2016. Enhanced terrestrial carbon preservation promoted by reactive iron in deltaic sediments. Geophysical Research Letters 43 (3): 1149–1157. https://doi.org/10.1002/2015GL067388.
Sigman, D.M., M.A. Altabet, R. Michener, D.C. McCorkle, B. Fry, and R.M. Holmes. 1997. Natural abundance-level measurement of the nitrogen isotopic composition of oceanic nitrate: an adaptation of the ammonia diffusion method. Marine Chemistry 57: 227–242. https://doi.org/10.1016/S0304-4203(97)00009-1.
Straub, K.L., M.- Benz, B. Schink, and F. Widdel. 1996. Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Applied and Environmental Microbiology 62: 1458–1460.
Sørensen, J., J.M. Tiedje, and R.B. Firestone. 1980. Inhibition by sulfide of nitric and nitrous oxide reduction by denitrifying Pseudomonas fluorescens. Applied and Environmental Microbiology 39 (1): 105–108.
van den Berg, E.M., J.L. Rombouts, J.G. Kuenen, R. Kleerebezem, and M.C. van Loosdrecht. 2017. Role of nitrite in the competition between denitrification and DNRA in a chemostat enrichment culture. AMB Express 7 (1): 91. https://doi.org/10.1186/s13568-017-0398-x.
Weber, K.A., L.A. Achenbach, and J.D. Coates. 2006. Microorganisms pumping iron: anaerobic microbial iron oxidation and reduction. Nature Rev Microbiol 4 (10): 752–764. https://doi.org/10.1038/nrmicro1490.
Acknowledgments
We thank members of the Paerl Lab, including B. Abare, J. Braddy, K. Rossignol, and R. Sloup, for assistance during the incubations and sample analysis. We also thank S. Jokinen (UEF) for assistance during the IRMS analyses and C. Treat (UEF) for help with the map.
Funding
Open access funding provided by University of Eastern Finland (UEF) including Kuopio University Hospital. This research was funded by the Academy of Finland (grant nos. 275127 and 307331 (Jäntti), 310302 (Aalto)) and joint funding by Olvi Foundation, Jenny and Antti Wihuri foundation, and Saastamoinen funding (Jäntti). The works were partially supported by the US National Science Foundation (DEB 1240851; OCE 1705972), The North Carolina Dept. of Environmental Quality and US National Fish and Wildlife Federation (Project 8020.16.053916: ModMon Program), the Lower Neuse Basin Association, NC Sea Grant, and the UNC Water Resources Research Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Marco Bartoli
Electronic Supplementary Material
ESM 1
(DOCX 211 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jäntti, H., Aalto, S.L. & Paerl, H.W. Effects of Ferrous Iron and Hydrogen Sulfide on Nitrate Reduction in the Sediments of an Estuary Experiencing Hypoxia. Estuaries and Coasts 44, 1–12 (2021). https://doi.org/10.1007/s12237-020-00783-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12237-020-00783-4