
Abstract
A loop-mediated isothermal amplification (LAMP) method with a real-time monitoring system was developed for the detection of porcine circovirus type 1 (PCV1) in commercial swine vaccines. This method was highly specific for PCV1. No cross-reaction to porcine circovirus type 2, porcine parvovirus, pseudorabies virus, classical swine fever virus, and porcine reproductive and respiratory syndrome virus was observed. The analytical sensitivity of the LAMP for PCV1 DNA was 10 copies/μl in the case of positive recombinant plasmid comparable to that obtained from the nested polymerase chain reaction (nested PCR). Furthermore, 25 commercial swine vaccines were tested by both the LAMP and the nested PCR, and three of them were tested positive for PCV1 DNA. These results indicate that PCV1 DNA can be real-time detected by the LAMP; the method was highly specific, sensitive, and rapid for the detection of PCV1 DNA, particularly in commercial swine vaccines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Porcine circovirus (PCV) belongs to the genus Circovirus of the family Circoviridae (Finsterbusch and Mankertz 2009). PCV is non-enveloped, 17 nm in size, and contains a circular single-stranded DNA genome with two major open reading frames, ORF1 and ORF2 (Finsterbusch and Mankertz 2009). ORF1 is believed to encode replication-associated proteins (Cheung 2003; Finsterbusch et al. 2005; Mankertz et al. 2004). ORF2 encodes a major capsid protein containing immunologically important domains associated with virus neutralization (Lekcharoensuk et al. 2004; Mahé et al. 2000). PCV has been divided into two distinct clusters, PCV1 and PCV2 (An et al. 2009; Calsamiglia et al. 2002; Cságola et al. 2008; Muhling et al. 2006). PCV2 can be distinguished from PCV1 on the basis of genome sequence and antigenicity (Calsamiglia et al. 2002; Mahe et al. 2000; Muhling et al. 2006).
PCV1 has been isolated from contaminated PK-15 cells (Dulac and Afshar 1989; Tischer et al. 1995) and is not considered to be pathogenic for pigs (Allan et al. 1995; Tischer et al. 1986). Presence of PCV1 in domestic pigs is not surprising because a number of viral vaccines are manufactured from porcine-derived components, which may be contaminated with PCV1 (Quintana et al. 2006). Meanwhile, contamination of PCV1 in vaccine products is unacceptable, and such products should not be used in pig farms (Quintana et al. 2006). PCV2, unlike PCV1, is pathogenic to pigs and is considered as the primary causative agent of post-weaning multisystemic wasting syndrome (PMWS), porcine dermatitis and nephropathy syndrome (PDNS), porcine respiratory disease complex (PRDC), proliferative and necrotizing pneumonia (PNP), and porcine circovirus-associated disease (PCVAD) (Chae 2005; Kixmöller et al. 2008; Wang et al. 2004). Recently, the emergence of PCV2 infection has led to a significant increase in post-weaning mortality and high economic losses for the swine industry around the world (Horlen et al. 2007; Kixmöller et al. 2008).
Loop-mediated isothermal amplification (LAMP) is an excellent diagnostic tool for detecting specific nucleic acid sequences in clinical samples (Alhassan et al. 2007; Chen et al. 2008; de Castro et al. 2008; Han et al. 2007; Notomi et al. 2000). This technique is highly specific, relatively fast, and easy to perform. In the LAMP reaction, a DNA sample is incubated with specific LAMP primer sets, Bst DNA polymerase with strand-displacement activity, and substrates at a constant temperature between 60°C and 65°C (Alhassan et al. 2007; Chen et al. 2008; Ihira et al. 2004; Nagashima et al. 2007). The LAMP has been used successfully to detect PCV2 in field samples (Chen et al. 2008), but has not yet been developed to detect PCV1 in commercial swine vaccines. The aim of this study was to develop a LAMP with a real-time monitoring system for the detection of PCV1 in commercial swine vaccines.
Materials and methods
Commercial porcine vaccines and viruses
A total of 25 commercially available porcine vaccines were obtained from licensed manufacturers and pharmaceutical suppliers in Taiwan (Table 1). Viral DNA was extracted from each vaccine using the QIAamp DNA Extraction Kit (Qiagen, USA) according to the manufacturer’s instructions. For evaluating specificity of the LAMP, 21 virus isolates, comprising six PCV1, five PCV2, two porcine parvovirus (PPV), three pseudorabies virus (PRV), three classic swine fever virus (CSFV), and two porcine reproductive and respiratory syndrome virus (PRRSV) isolates, were used; all of the isolates were previously identified by determining their partial nucleotide sequences (Wang et al. 2004, 2008; Huang et al. 2009). The DNA of PCV1, PCV2, PPV, PRV, and cDNA of CSFV and PRRSV isolates were produced as described previously (Huang et al. 2009; Wang et al. 2004, 2008)
Primers
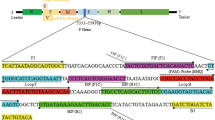
Specific primer sets of the LAMP for PCV1 detection were designed by PrimerExplorer V4 software (http://primerexplorer.jp/elamp4.0.0/index.html) based on different reference PCV1 isolates (GenBank accession no. AF012107, AY184287, AY219836, AY660574, DQ472013, DQ659154, EF493843, FJ475129, and GU722334). The primer sets consisted of two outer primers (F3 and B3) and two inner primers (FIP and BIP) targeting a conserved region within the PCV1 ORF2 gene. To further validate the LAMP, the primers PCV-F1, PCV-R1, PCV-F2, and PCV-R2 were used for nested polymerase chain reaction (nested PCR) targeting the conserved region of the PCV1 ORF2 gene (Victoria et al. 2010). Nucleotide sequences and locations of the primers are shown in Table 2.
LAMP reaction
A 25-μl reaction mixture consisted of 12.5 μl of 2× LAMP reaction buffer (Eiken Chemical, Japan), 8 U Bst DNA polymerase (New England Biolabs, USA), 2 μl of the extracted target DNA sample, 40 pmol each of FIP and BIP primers, and 5 pmol each of F3 and B3 primers. The LAMP reaction was conducted by incubating the reaction mixture at 60°C for 60 min and then inactivating the Bst DNA polymerase at 80°C for 2 min. The reaction was monitored in real-time at 6-s intervals by measuring the turbidity at A650 using a Loopamp real-time turbidimeter (LA-320; Eiken Chemical, Japan). The cutoff turbidity was set at 0.1 turbidity units and was determined by analyzing negative controls in replicates. A sample with turbidity remaining below 0.1 turbidity units throughout the 60-min incubation period was considered to be negative for PCV1 DNA. If the turbidity of a sample increased above 0.1 turbidity units within the 60-min incubation period, it was considered to be positive, indicating contamination with PCV1 DNA. The LAMP amplicons were further analyzed by electrophoresis through a 2% agarose gel containing 0.5 mg/ml SYBR Safe DNA gel stain (Invitrogen, USA) in Tris–acetate–EDTA (ethylenediamine tetra-acetic acid) buffer.
Nested PCR reaction
A total of 25 PCR reaction mixtures of the first round PCR in the nested PCR consisted of 2.5 μl of 10× buffer (100 mmol/l Tris–HCl at pH 8.8, 500 mmol/l KCl, 15 mmol/l MgCl2, 1% Triton X-100), 1.25 μmol/l each of four nucleoside triphosphates, 20 μmol/l of each primer, 0.5 μl of DNA polymerase POWER TAQ (2 U/μl; Bertec, Taipei, Taiwan), and 2 μl of the DNA sample. The first round PCR was performed at 95°C for 5 min, followed by 30 cycles of denaturation at 95°C for 20 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s. Finally, the samples were subjected to a terminal extension step at 72°C for 10 min. From this reaction, 2 μl of the first round PCR product was used as a template for the nested PCR. In the nested PCR, 25 μl of reaction mixture consisted of 2.5 μl of 10× buffer (100 mmol/l Tris–HCl at pH 8.8, 500 mmol/l KCl, 15 mmol/l MgCl2, 1% Triton X-100), 1.25 μmol/l each of four nucleoside triphosphates, 20 μmol/l of each primer, and 0.5 μl of DNA polymerase POWER TAQ (2 U/μl; Bertec, Taiwan). The nested PCR was performed at 95°C for 5 min, followed by 30 cycles each of denaturation at 95°C for 20 s, annealing at 55°C for 20 s, and extension at 72°C for 20 s. Finally, the samples were subjected to a terminal extension step at 72°C for 10 min. Amplicon of the nested PCR were analyzed by electrophoresis through a 2% agarose gel as described above.
Construction of recombinant plasmids
Full-length ORF2 gene of PCV1 (GenBank accession no. AY219836) was amplified as described previously (Lekcharoensuk et al. 2004). The amplicon was ligated into the pCRII-TOPO plasmid that was supplied with the Dual Promoter TOPO TA Cloning Kit (Invitrogen, USA). Top10F® Escherichia coli competent cells (Invitrogen, USA) were transformed with the recombinant plasmid according to the manufacturer’s instructions. The inserted target gene carried by the recombinant plasmid was directly sequenced as described previously (Wang et al. 2004). The recombinant plasmid was quantified by measuring its absorbance A 260 on a U2100 pro UV/Vis spectrophotometer (General Electronic Healthcare, Singapore) and diluted to 107 copies/μl to develop a standard curve for evaluating the analytical sensitivity of the LAMP.
Analytical specificity and sensitivity of the LAMP
To determine the analytical specificity of the LAMP, DNA samples extracted from the 21 virus isolates were tested by the LAMP. DNA extracted from specific-pathogen-free (SPF) swine tissue samples was used as the negative control. To evaluate the analytical sensitivity of the assay, 10-fold serial dilutions of the recombinant plasmid were performed to prepare the dilutions at concentration from 107 copies/μl to 1 copy/μl, and each dilution was tested by both the LAMP and the nested PCR (Victoria et al. 2010). For quantification of the diluted samples, a standard curve was generated for the LAMP, with serial 10-fold dilutions of the recombinant plasmid that were positive for PCV1 DNA on the X-axis and the time of positivity (min) on the Y-axis.
Evaluation of LAMP with commercial swine vaccines
Two batches of each vaccine were tested for detection of PCV1 DNA by the LAMP and the nested PCR, respectively. To evaluate the vaccine matrices for interference or inhibition of the LAMP reaction, all extracted DNA of tested vaccine samples were spiked with plasma containing the recombinant plasmid. The final concentration of the recombinant plasmid for each spiked vaccine sample was 106 copies/ml. Detection of PCV1 DNA for each spiked vaccine sample was performed by the LAMP as an internal control.
Results
The analytical specificity of LAMP was evaluated using DNA extracted from PCV1, PCV2, PPV, and PRV, and cDNA derived from CSFV and PRRSV. PCV1 DNA was successfully detected, and a ladder-like amplicon was generated (Fig. 1). The LAMP amplified the PCV1 target gene, but not the nucleic acids extracted from PCV2, PPV, PRV, CSFV, and PRRSV (Fig. 1).

Analytical specificity of the LAMP was observed using agarose gel electrophoresis. Lane M 2,000–100 bp ladder marker (50 U/μl; Protech, Taiwan); lanes 1–7 extracted DNA of PCV1, PCV2, PPV, PRV, cDNA of CSFV, PRRSV, and negative control, respectively
By testing 10-fold serial dilutions (107 copies/μl to 1 copy/μl) of the recombinant plasmid, the LAMP demonstrated a detection limit of 10 copies (Fig. 2, lane 7), comparable to the nested PCR (10 copies; Fig. 2, lane 15). The standard curve for LAMP was generated from the amplification plots of the 10-fold serial dilutions of the recombinant plasmids (107 copies/μl to 10 copies/μl) vs. time (min) (Fig. 3a). A linear regression line with a coefficient of r 2 = 0.97 was plotted for the LAMP (Fig. 3b).

Analytical sensitivities of the LAMP (lanes 1–8) and the nested PCR (lanes 9–16) were compared by detecting 10-fold serial dilutions of the recombinant plasmid (107 copies/μl to 1 copy/μl) by agarose gel electrophoresis. Lane M 2,000–100 bp ladder marker (50 U/μl; Protech, Taiwan). The PCR products of lanes 9–15 were 155 base pairs (bp) in length

Analytical sensitivity of the LAMP (a) was performed by real-time monitoring with 10-fold serial dilutions of the recombinant plasmid: 107 copies/μl to 1 copy/μl and negative control (NT). The linear function fitted to the plot of the mean threshold time against the log of the input PCV1 the recombinant plasmid is also shown (b)
PCV1 DNA was detected in three commercial CSFV vaccines by the LAMP and the nested PCR (Table 2). These vaccines were manufactured by three different commercial companies (B, C, and E). All extracted DNA of tested vaccine samples were spiked with PCV1 recombinant plasmids and yielded positive results for detection of PCV1 DNA by the LAMP (data not shown). These results also indicated that inhibition was not observed in the extracted DNA of 25 commercial swine vaccine samples.
Discussion
The LAMP developed in this study was rapid for the detection of PCV1 DNA. The products generated by this method can be analyzed not only by conventional agarose gel electrophoresis but also spectrophotometry, which enables real-time analysis and quantification of the amplicons in commercial swine vaccines. Amplification of a specific genomic region by conventional or nested PCR alone, or by PCR in combination with gene sequencing, has long been used to confirm the accuracy of amplicons generated by PCR (Muhling et al. 2006; Victoria et al. 2010). However, with or without gene sequencing, conventional detection procedures for PCV1 require several hours to complete. In contrast, DNA detection with the LAMP was much faster than with conventional PCR or nested PCR. After preparation of the sample DNA, the LAMP could be completed in approximately 1 h, whereas other methods required several hours to a few days.
Furthermore, PCV1 DNA could be specifically and sensitively amplified using the two LAMP primer sets; no amplification was observed when the method was used to detect nucleic acids of PCV2, PPV, PRV, CSFV, FMDV, and PRRSV. These results indicate the specificity and wide applicability of our LAMP method in the detection of viral contaminants in commercial swine vaccines. The sensitivity of the LAMP was equivalent to that of the nested PCR. Our results showed that the LAMP is highly sensitive and can detect small amounts of PCV1 DNA present in samples. This method enables the detection and quantification of PCV1 DNA in commercial swine vaccines.
In this study, using the LAMP, we found that three commercially available CSFV vaccines were contaminated with PCV1 DNA, although the source of PCV1 DNA contamination in some commercial porcine vaccines has remained unknown. Recently, PCV1 DNA was also found in two commercial human rotavirus vaccines (Baylis et al. 2011; Victoria et al. 2010). Although PCV1 is not regarded as a potential pathogen in humans (Victoria et al. 2010), avoiding virus contamination during the process of vaccine production is absolutely essential. The detected PCV1 DNA may originate from the cell lines used for vaccine production (Quintana et al. 2006). Presence of PCV1 DNA in the vaccines does not necessarily imply that infectious PCV1 particles are present in the vaccines. The contaminating PCV1 may have been inactivated during the manufacturing process or may have been present in the live vaccine products in too small amounts to cause an infection (Quintana et al. 2006). Regardless of whether the contaminant PCV1 was alive or inactivated, the presence of viral DNA other than the vaccine DNA itself is absolutely unacceptable. To fulfill this requirement, a rapid, specific, and sensitive method such as LAMP can be easily incorporated in the production process to screen for PCV1 contamination in vaccines and in cell lines used for cultivation of the vaccine viruses.
The spiking study was performed by deliberately spiking PCV1 virus into the vaccine samples to account for inhibition; this avoids a potential problem that may result in the under-detection of PCV1 (i.e., false negatives). A previous study indicated that some human medicines contain heparin, adjuvant, phenol, or formaldehyde, which are common PCR inhibitors and always remain with the extracted nuclear acid (Yokota et al. 1999). In this study, no evidence of inhibition has been observed in any extracted DNA of swine vaccine sample for spiked recombinant plasmids. However, this was necessary to perform spiking studies as an internal control for the LAMP reaction; this assessed individual vaccine matrices that may be the cause of amplification product inhibition.
In conclusion, our LAMP method enables the detection of small amounts of PCV1 DNA. This method can be used to rapidly detect cell lines and commercial swine vaccines for PCV1 contamination with high sensitivity and specificity.
Abbreviations
- PCV1:
-
Porcine circovirus type 1
- PCV2:
-
Porcine circovirus type 2
- PMWS:
-
Post-weaning multisystemic wasting syndrome
- PDNS:
-
Porcine dermatitis and nephropathy syndrome
- PCVAD:
-
Porcine circovirus associated disease
- PNP:
-
Proliferative and necrotizing pneumonia
- ORF:
-
Open reading frame
- LAMP:
-
Loop-mediated isothermal amplification
- PCR:
-
Polymerase chain reaction
- PPV:
-
Porcine parvovirus
- PRRSV:
-
Porcine reproductive and respiratory syndrome virus
- CSFV:
-
Classic swine fever virus
- PRV:
-
Pseudorabies virus
References
An D, Song DS, Park JY, Park BK (2009) A DNA miniarray system for simultaneous visual detection of porcine circovirus type 1 (PCV1) and 2 (PCV2) in pigs. Vet Res Commun 33:139–147
Alhassan A, Thekisoe OM, Yokoyama N, Inoue N, Motloang MY, Mbati PA, Yin H, Katayama Y, Anzai T, Sugimoto C, Igarashi I (2007) Development of loop-mediated isothermal amplification (LAMP) method for diagnosis of equine piroplasmosis. Vet Parasitol 143:155–160
Allan GM, McNeilly F, Cassidy JP, Reilly GA, Adair B, Ellis WA, McNulty MS (1995) Pathogenesis of porcine circovirus: experimental infections of colostrum deprived piglets and examination of pig foetal material. Vet Microbiol 44:49–64
Baylis SA, Finsterbusch T, Bannert N, Blümel J, Mankertz A (2011) Analysis of porcine circovirus type 1 detected in Rotarix vaccine. Vaccine 29:690–697
Calsamiglia M, Segalés J, Quintana J, Rosell C, Domingo M (2002) Detection of porcine circovirus types 1 and 2 in serum and tissue samples of pigs with and without postweaning multisystemic wasting syndrome. J Clin Microbiol 40:1848–1850
Chae C (2005) A review of porcine circovirus 2-associated syndromes and diseases. Vet J 169:326–336
Chen HT, Zhang J, Sun DH, Chu YF, Cai XP, Liu XT, Luo XN, Liu Q, Liu YS (2008) Rapid detection of porcine circovirus type 2 by loop-mediated isothermal amplification. J Virol Methods 149:264–268
Cheung AK (2003) Transcriptional analysis of porcine circovirus type 2. Virology 305:168–180
Cságola A, Kiss I, Tuboly T (2008) Detection and analysis of porcine circovirus type 1 in Hungarian wild boars: short communication. Acta Vet Hung 56:139–144
de Castro AM, Cortez A, Heinemann MB, Brandão PE, Richtzenhain LJ (2008) Molecular diversity of Brazilian strains of porcine circovirus type 2 (PCV-2). Res Vet Sci 85:197–200
Dulac GC, Afshar A (1989) Porcine circovirus antigens in PK-15 cell line (ATCC CCL-33) and evidence of antibodies to circovirus in Canadian pigs. Can J Vet Res 53:431–433
Finsterbusch T, Steinfeldt T, Caliskan R, Mankertz A (2005) Analysis of the subcellular localization of the proteins Rep, Rep’ and Cap of porcine circovirus type 1. Virology 343:36–46
Finsterbusch T, Mankertz A (2009) Porcine circoviruses—small but powerful. Virus Res 143:177–183
Han ET, Watanabe R, Sattabongkot J, Khuntirat B, Sirichaisinthop J, Iriko H, Jin L, Takeo S, Tsuboi T (2007) Detection of four Plasmodium species by genus- and species-specific loop-mediated isothermal amplification for clinical diagnosis. J Clin Microbiol 45:2521–2528
Horlen KP, Schneider P, Anderson J (2007) A cluster of farms experiencing severe porcine circovirus associated disease: clinical features and association with the PCV2b genotype. J Swine Health Prod 15:270–278
Huang YL, Pang VF, Pan CH, Chen TH, Jong MH, Huang TS, Jeng CR (2009) Development of a reverse transcription multiplex real-time PCR for the detection and genotyping of classical swine fever virus. J Virol Methods 160:111–118
Ihira M, Yoshikawa T, Enomoto Y, Akimoto S, Ohashi M, Suga S, Nishimura N, Ozaki T, Nishiyama Y, Notomi T, Ohta Y, Asano Y (2004) Rapid diagnosis of human herpesvirus 6 infection by a novel DNA amplification method, loop-mediated isothermal amplification. J Clin Microbiol 42:140–145
Kixmöller M, Ritzmann M, Eddicks M, Saalmüller A, Elbers K, Fachinger V (2008) Reduction of PMWS-associated clinical signs and co-infections by vaccination against PCV2. Vaccine 25:3443–3451
Lekcharoensuk P, Morozov I, Paul PS, Thangthumniyom N, Wajjawalku W, Meng XJ (2004) Epitope mapping of the major capsid protein of type 2 porcine circovirus (PCV2) by using chimeric PCV1 and PCV2. J Virol 78:8135–8145
Mankertz A, Caliskan R, Hattermann K, Hillenbrand B, Kurzendoerfer P, Mueller B, Schmitt C, Steinfeldt T, Finsterbusch T (2004) Molecular biology of Porcine circovirus: analyses of gene expression and viral replication. Vet Microbiol 98:81–88
Mahe D, Blanchard P, Truong C, Arnauld C, Le Cann P, Cariolet R, Madec F, Albina E, Jestin A (2000) Differential recognition of ORF2 protein from type 1 and type 2 porcine circoviruses and identification of immunorelevant epitopes. J Gen Virol 81:1815–1824
Muhling J, Raye WS, Buddle JR, Wilcox GE (2006) Genetic characterisation of Australian strains of porcine circovirus types 1 and 2. Aust Vet J 84:421–425
Nagashima S, Yoshida A, Ansai T, Watari H, Notomi T, Maki K, Takehara T (2007) Rapid detection of the cariogenic pathogens Streptococcus mutans and Streptococcus sobrinus using loop-mediated isothermal amplification. Oral Microbiol Immuno 22:361–368
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T (2000) Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 15:e63
Quintana J, Segalés J, Calsamiglia M, Domingo M (2006) Detection of porcine circovirus type 1 in commercial pig vaccines using polymerase chain reaction. Vet J 171:570–573
Tischer I, Mields W, Wolff D, Vagt M, Griem W (1986) Studies on epidemiology and pathogenicity of porcine circovirus. Arch Virol 91:271–276
Tischer I, Bode L, Peters D, Pociuli S, Germann B (1995) Distribution of antibodies to porcine circovirus in swine populations of different breeding farms. Arch Virol 140:737–743
Victoria JG, Wang C, Jones MS, Jaing C, McLoughlin K, Gardner S, Delwart EL (2010) Viral nucleic acids in live-attenuated vaccines: detection of minority variants and an adventitious virus. J Virol 84:6033–6040
Wang C, Huang TS, Huang CC, Tu C, Jong MH, Lin SY, Lai SS (2004) Characterization of porcine circovirus type 2 in Taiwan. J Vet Med Sci 66:469–475
Wang C, Lee F, Huang TS, Pan CH, Jong MH, Chao PH (2008) Genetic variation in open reading frame 5 gene of porcine reproductive and respiratory syndrome virus in Taiwan. Vet Microbiol 131:339–347
Yokota M, Tatsumi N, Nathalang O, Yamada T, Tsuda I (1999) Effects of heparin on polymerase chain reaction for blood white cells. J Clin Lab Anal 13:133–140
Acknowledgments
The authors are grateful to those who submitted commercial porcine vaccines to Division of Hog Cholera Research, Animal Health Research Institute, Executive Yuan. The authors also thank Dr. Richard Huang and Dr. Eric Wang (Protech Technology Enterprise Co., Ltd., Taipei, Taiwan) for their excellent technical help.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wang, C., Pang, V.F., Jeng, CR. et al. Detection of porcine circovirus type 1 in commercial porcine vaccines by loop-mediated isothermal amplification. Folia Microbiol 56, 483–489 (2011). https://doi.org/10.1007/s12223-011-0072-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12223-011-0072-7