
Abstract
Water electrolysis, a process for producing green hydrogen from renewable energy, plays a crucial role in the transition toward a sustainable energy landscape and the realization of the hydrogen economy. Oxygen evolution reaction (OER) is a critical step in water electrolysis and is often limited by its slow kinetics. Two main mechanisms, namely the adsorbate evolution mechanism (AEM) and lattice oxygen oxidation mechanism (LOM), are commonly considered in the context of OER. However, designing efficient catalysts based on either the AEM or the LOM remains a topic of debate, and there is no consensus on whether activity and stability are directly related to a certain mechanism. Considering the above, we discuss the characteristics, advantages, and disadvantages of AEM and LOM. Additionally, we provide insights on leveraging the LOM to develop highly active and stable OER catalysts in future. For instance, it is essential to accurately differentiate between reversible and irreversible lattice oxygen redox reactions to elucidate the LOM. Furthermore, we discuss strategies for effectively activating lattice oxygen to achieve controllable steady-state exchange between lattice oxygen and an electrolyte (OH− or H2O). Additionally, we discuss the use of in situ characterization techniques and theoretical calculations as promising avenues for further elucidating the LOM.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The development of methods for hydrogen production from water electrolysis provides a key technical strategy for the large-scale usage of renewable energy in future [1]. In water electrolysis, the cathodic hydrogen evolution reaction is a two-electron transfer process, and the anodic oxygen evolution reaction (OER) involves a complex four-electron transfer process. This results in slow reaction kinetics and high overpotential, which are the main factors leading to high energy consumption [2,3,4,5]. To obtain high-performance OER catalysts, researchers have conducted long-term and in-depth exploration, leading to crucial breakthroughs in elucidating the catalytic reaction mechanisms. The adsorbate evolution mechanism (AEM) and the lattice oxygen oxidation mechanism (LOM) are the main mechanisms of OER. It has been demonstrated that the minimum theoretical overpotential for the AEM is 0.37 V, while LOM can overcome this limitation and further reduce the theoretical overpotential [2, 6]. However, from the thermodynamic perspective, the LOM involves the participation of surface lattice oxygen in the reaction, which may lead to catalyst instability [7]. Numerous investigations have been conducted to determine the prospect of designing a catalyst with high activity and stability by modifying the OER mechanism [2, 8]. Lu et al. [9] achieved controllable conversion of the OER mechanism from AEM-to-LOM-to-AEM by precisely regulating the oxygen defect content in LaxSr1−xCoO3−δ, which provides motivations for achieving synergistic optimization of catalyst activity and stability by altering the mechanism rather than focusing on the specific design of catalysts. In addition, future research emphasis will be placed on the development of OER catalysts driven by either the AEM or LOM in acidic environments as these conditions often present more significant challenges in terms of activity and stability [10, 11]. Therefore, a comprehensive understanding of the OER mechanism is crucial for the development of efficient electrocatalysts for water oxidation. Based on the current research reports and comparing the characteristics, advantages, and disadvantages of the AEM and LOM, we propose insights into developing OER catalysts based on the LOM in future.
Mainstream Reaction Mechanism
There are two main reaction mechanisms of OER: (1) AEM, in which a metal acts as the redox center, and the electronic state near the Fermi level exhibits metallic characteristics; (2) LOM, in which oxygen acts as the redox center, and the electronic state near the Fermi level exhibits oxygen-like characteristics [12]. In the traditional AEM, the OER involves various reaction intermediates, including *OH, *O, *OOH, and *O2, which are adsorbed on metal active centers. The minimum theoretical overpotential for the investigated catalysts based on AEM is predicted to be ~ 0.37 V resulting from a linear scaling relation between the adsorption energies of *OH and *OOH intermediates (ΔG*OOH = ΔG*OH + 3.2 eV). However, this scaling relation cannot explain some reported catalysts with lower overpotentials [2, 13, 14]. The presence of such a scaling relation imposes limitations on the development of catalysts designed to approach the equilibrium potential of the OER. However, this scaling relation also offers certain advantages. For instance, the scaling relation-based volcano plot can serve as a universal tool for predicting catalyst activity (in accordance with the Sabatier principle), thereby reducing experimental and computational costs [15]. Additionally, electrocatalysts based on the AEM, which is characterized by the reaction path, demonstrated relatively high stability [7].
However, to further reduce the theoretical overpotential for the OER, it is necessary to overcome the inherent scaling relation between the adsorption energies of *OH and *OOH. Therefore, researchers have proposed an alternative mechanism known as the LOM, in which oxygen ligands are activated and released from the lattice matrix during the electrochemical reactions. This subsequent reaction process enables the direct coupling of O–O radicals and is believed to overcome the aforementioned limitation [16, 17]. In particular, the adsorbed OH− undergoes deprotonation to form O2−, and electrons from the oxygen orbital of O2− are transferred to the external circuit, eventually forming radical O− species (a nonbonding state of oxygen) in which oxygen acts as the redox center. Subsequently, the oxygen nonbonding states in adjacent O− atoms hybridize to form (O–O)2p bands, thereby overcoming the limitation imposed by the scaling relations. Consequently, catalytic activity in the LOM is typically higher than that in the traditional AEM. Furthermore, most studies have indicated that, unlike the AEM, the transfer process of electrons and protons in the LOM is decoupled, rendering the OER process pH dependent [16, 18]. Moreover, unlike the more stable metal active center in the AEM, the lattice oxygen in the catalyst structure and the oxygen in the electrolyte (OH− or H2O) exist in a dynamic exchange state in the LOM since oxygen serves as the active center. Although the LOM can greatly enhance the OER activity, it may affect the catalyst’s stability when the lattice oxygen in the catalyst overflows excessively.
Different Forms of Lattice Oxygen Oxidation Reaction
Some researchers may misunderstand the LOM, as lattice oxygen oxidation reactions can be categorized into reversible oxygen redox reactions and irreversible oxygen redox reactions [8]. Conducting an in-depth analysis of these two forms of lattice oxygen oxidation is crucial to avoid misconceptions regarding the LOM. In the reversible LOM pathway, the activation of lattice oxygen is a prerequisite, followed by specific oxidation, exchange, and release processes of lattice oxygen ligands on the catalyst surface. Once the LOM pathway is successfully initiated, the OH/H2O group binds to the oxygen vacancy, acting as a regenerated “lattice oxygen” for the subsequent cycle. However, the catalyst surface will change dynamically, which may lead to structural instability, often referred to as the “instantaneous dissolution mechanism” [19]. Thus, the involvement of lattice oxygen may also reduce the catalyst stability. The irreversible LOM pathway involves a much higher overflow rate of lattice oxygen on the material surface compared with the filling rate of OH− in the electrolyte. This leads to the generation of numerous oxygen vacancies on the catalyst surface. The generation of vacancies is often accompanied by the severe leaching of metal cations, resulting in irreversible surface reconstruction and the formation of metal oxyhydroxide. The surface reconstruction caused by this irreversible lattice oxygen oxidation disrupts the crystal structure of the catalyst, turning it into the precursor or precatalyst of the actual active species. Furthermore, owing to the absence of oxygen nonbonding states on the reconstructed oxyhydroxide surface, the subsequent OER process may follow the AEM rather than the LOM. Therefore, the aforementioned true advantages of LOM may not be realized [7, 8].
These two activation forms lead to essential differences in surface reactions, but they also share commonalities. The presence of oxygen orbitals near the Fermi level is a prerequisite for triggering the redox activity of oxygen. The closer the position of the O 2p band is to the Fermi level, the more favorable it is for oxygen ion diffusion to form oxygen vacancies and release O2. Although the LOM pathway can overcome the overpotential limitation of AEM, the deprotonation process becomes significantly more challenging owing to the transition of the active center from delocalized metal orbitals to localized oxygen nonbonding orbitals. Consequently, there are instances in which LOM-based catalysts do not exhibit superior activity to AEM-based catalysts [20].
Origin and Regulation of Lattice Oxygen Oxidation Mechanism
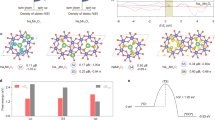
As mentioned earlier, lattice oxygen possesses a unique electronic structure, and understanding the chemical and structural origin of lattice oxygen oxidation is crucial in elucidating the LOM. We used transition metal hydroxides and alkali metal oxides as model catalysts to investigate the chemical and structural origin of the advanced LOM of water oxidation. Notably, O–O bonds can be formed through two pathways: acid–base nucleophilic attack and direct O–O coupling [21]. The acid–base nucleophilic attack pathway is subject to the scaling relation because it involves the formation of *OOH. However, in the O–O direct coupling pathway, O2 can be directly generated without the involvement of *OOH, thereby overcoming the limitation imposed by the scaling relation. For instance, we promoted the formation of nonbonded oxygen (ONB) states by incorporating low-valence, inert Zn2+ (d10) ions in CoOOH (Fig. 1a). The presence of oxygen holes, which is direct evidence of oxygen oxidation, indicates that the lattice oxygen oxidation chemically originates from the unstable oxygen electrons extracted from ONB. Moreover, oxygen holes in ONB can facilitate O–O direct coupling and nucleophilic OH− attack. In the case of pure CoOOH, the traditional AEM pathway dominates due to the absence of nonbonded oxygen states. Upon introducing 10% Zn2+, the nucleophilic attack path of the lattice oxygen mechanism occurs (following the scaling relation). By further increasing the amount of Zn2+ to 20%, the O–O direct coupling pathway can be activated, thereby overcoming the limitation of the scaling relation. However, excessive doping of Zn2+ (50%) may result in unfavorable energetics for oxygen vacancy refilling in Zn0.5Co0.5OOH, leading to severe degradation. Importantly, for O–O direct coupling to occur in the LOM, it is crucial that two neighboring oxidized oxygen atoms rotate and hybridize their holes without significantly compromising the metal–oxygen hybridization. In cases where this condition is not met, the acid–base nucleophilic attack pathway promoted by lattice oxygen will occur, which remains constrained by the scaling relation.

Copyright 2019 Springer Nature. Panels b and c were reproduced with permission from Ref. [22]. Copyright 2021 Springer Nature
a Transition from adsorbate evolution mechanism to lattice oxygen oxidation mechanism. b Schematic of oxygen hole formation in oxygen lone-pair states (|O2p) for NaxMn3O7. c Schematic of the oxygen evolution reaction pathway of acid–base nucleophilic attack. Panel a was reproduced with permission from Ref. [17].
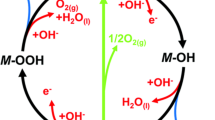
To further overcome the theoretical overpotential limitation imposed by the acid–base nucleophilic attack pathway, we present an effective strategy for preparing improved oxygen evolution electrocatalysts by adjusting the lattice oxygen reactivity and the scaling relation through alkali metal ion mediation using modeled NaxMn3O7 materials (Fig. 1b) [22]. The NaxMn3O7 structure features nonbonded oxygen states owing to the presence of native Mn vacancies, and reducing the Na+ content can activate lattice oxygen for the LOM. However, this model lacks the structural characteristics that promote O–O direct coupling. The oxygen holes may trigger the formation of O–O bonds through an enhanced acid–base nucleophilic attack, in which the deprotonation of *OH involves only proton transfer (M–OH + OH− → M–O− + H2O) and is decoupled during subsequent oxygen release (Fig. 1c). Additionally, Na+ can interact with the pendant oxygen of *OOH through specific noncovalent interactions, which further stabilizes *OOH without affecting the adsorption of *OH, thereby breaking the constraint imposed by the scaling relation. These results demonstrate that the predicted NaMn3O7 serves as the optimal electrocatalyst in this model by effectively adjusting the reactivity and proportion of lattice oxygen. These findings are strongly supported by experimental and characterization results.
There have been notable advancements in the lattice oxygen mechanism. For instance, researchers have proposed the lattice oxygen-mediated mechanism–oxygen vacancy site mechanism (LOM–OVSM) in the acidic OER system. Wang et al. created Ru–O–Rh active sites by doping Rh and introducing oxygen vacancies on the surface of RuO2. Their study revealed the optimal reaction pathway of LOM–OVSM induced by Ru–O–Rh sites rich in oxygen vacancies, thereby breaking the thermodynamic energy barrier limit of the traditional AEM (Fig. 2a, b) [23]. Additionally, Yao et al. [24] employed Ru–N bonding to anchor Ru oxide on UiO-67-bpydc at the atomic level, enhancing the covalency of the Ru–O bond and reducing the energy barrier required for lattice oxygen oxidation, resulting in ultrahigh activity and stability for acidic OER (Fig. 2c). Interestingly, Wang et al. [12] recently reported a novel coupled oxygen evolution mechanism (COM), which combines the respective advantages of AEM and LOM. In the COM, the catalytic reaction involves alternating redox centers between metal and oxygen: The metal acts as the redox center during deprotonation, while oxygen serves as the redox center during O–O bonding (Fig. 2d). This approach prevents the excessive dissolution of metals in the LOM and accelerates the deprotonation process, offering valuable insights for further exploration of advanced OER mechanisms based on the LOM.

Copyright 2023 Springer Nature. Panel c was reproduced with permission from Ref. [24]. Copyright 2023 Elsevier Inc. Panel d was reproduced with permission from Ref. [12]. Copyright 2022 Springer Nature
a Lattice oxygen mediated mechanism–oxygen vacancy site mechanism of Rh-RuO2. b A two-dimensional activity map for lattice oxygen mediated mechanism–oxygen vacancy site mechanism of Rh-RuO2. c Increased covalency of the Ru–O bond lowers the energy barrier for the lattice oxygen oxidation mechanism pathway. d Origins of the coupled evolution mechanism. Panels a and b were reproduced with permission from Ref. [23].
Directions to Develop Lattice Oxygen Oxidation Mechanism
A catalyst is often evaluated based on its activity and stability. This criterion is also crucial in determining the practical feasibility of the LOM. With advancements in advanced characterization techniques and theoretical calculations, researchers have gained a deeper understanding of the microscopic reaction mechanism involved in the OER. For instance, the observation of oxygen exchange between the catalyst and electrolyte through in situ Raman spectroscopy and online differential electrochemical mass spectrometry has provided direct experimental evidence for the LOM [16, 24]. Although the LOM has surpassed the theoretical overpotential limit of the traditional AEM, the resulting stability issue has emerged as a new challenge [7]. Therefore, the question of whether the LOM can effectively replace the AEM in designing more efficient and stable catalysts has become a topic of debate among researchers. Based on the current reports, a “steady-state exchange” process of lattice oxygen can be realized through chemical and physical regulation via the rational control of crystallinity, introduction of appropriate metal vacancies, and assistance of external light or magnetic field [12, 25,26,27,28,29]. Particularly, addressing the following key issues could lead to a significant breakthrough in the OER mechanism:
-
1.
Effective activation of lattice oxygen has long been a bottleneck in LOM development. The high electronegativity of oxygen renders the redox of lattice oxygen thermodynamically unfavorable. Enhancing the d–p orbital hybridization of metal sites with neighboring oxygen can aid lattice oxygen activation. For instance, using a single atom-supported catalyst may be a promising approach [25]. Through an ingenious design, active lattice oxygen can be confined around the single atom, preventing excessive overflow. Additionally, lattice oxygen can be activated through anionic ligand modification, such as halogen element doping and flexible ligand modification of active sites using MOF- and COF-based materials [30,31,32].
-
2.
Achieving stable exchange between lattice oxygen and electrolyte (OH− or H2O) is another major challenge. Ensuring a stable exchange of lattice oxygen with the reaction medium is essential for a catalyst to exhibit high stability. Moreover, maintaining an equilibrium between surface oxygen vacancy formation and the OH− filling rate is crucial to prevent uncontrollable surface reconstruction caused by excessive dissolution of metal cations. Moreover, only in this case can the LOM occur stably after the driving surface reconstruction, rather than the AEM. Unfortunately, as far as we know, there is no effective solution to this problem yet. However, optical/thermal/magnetic assistance may prove to be an important method in future for addressing this challenge.
-
3.
Enhancing the utilization of in situ characterization experiments to reveal the catalytic process of lattice oxygen at the reaction interface is essential. Techniques such as in situ spectroscopy coupled with chemical probe technology and isotope labeling technology can be powerful tools for identifying reaction intermediates. Additionally, in situ observation of the dynamic structural evolution during the reaction process is crucial. The activation of lattice oxygen can induce a metastable state on the surface, which is key to unraveling the real active sites after surface reconstruction resulting from lattice oxygen oxidation.
-
4.
Advancing the realistic utilization of theoretical calculation tools to elucidate the reaction mechanism is essential. Real catalytic reactions often occur at the catalyst surface–interface. However, numerous studies have primarily focused on the bulk phase, such as the d-band center, and proposed activity descriptors that are often based on the bulk phase structure. There is currently a pressing need for more profound surface–interface activity descriptors and more realistic reaction mechanisms. These advancements will provide strong theoretical guidance for the further development of the OER mechanism.
-
5.
Advancing the application of catalysts based on the LOM in industrial membrane electrode devices is another crucial aspect. Considering the practical application of the OER, in addition to using the LOM mechanism to design more efficient catalysts, the mechanism should be considered in the application of membrane electrode devices, such as proton exchange membrane water electrolysis under acidic conditions and anion exchange membrane water electrolysis in alkaline media. In addition to designing proton exchange membranes or anion exchange membranes with high ionic conductivity, mechanical properties, and stability, the development of membrane electrode assemblies with high-temperature resistance and high current is also crucial for achieving efficient water electrolysis. For instance, in the membrane electrode assembly preparation, it is important to address the urgent need for optimizing catalyst loading and adhesion to the substrate.
Conclusions
In recent years, considerable progress has been made in elucidating the LOM, including its chemical and structural origins and the development of regulating strategies. Herein, we highlight the advantages and disadvantages of LOM and AEM and emphasize the importance of addressing the compatibility of high activity and stability in LOM research. A comprehensive understanding of reversible and irreversible forms of lattice oxygen oxidation is crucial to avoid misconceptions regarding the LOM. Achieving a balance between the activation rate of lattice oxygen and the filling rate of the electrolyte (OH− or H2O) is the key to ensuring the reliable occurrence of the LOM pathway, and this requires further exploration in future. Additionally, we identified the need for advancements in situ characterization techniques and high-performance computing to more accurately elucidate the reaction mechanisms occurring at catalyst surfaces and interfaces. Furthermore, promoting the application of LOM-based catalysts in industrial membrane electrode devices is a promising direction for future research and development.
References
Du NY, Roy C, Peach R et al (2022) Anion-exchange membrane water electrolyzers. Chem Rev 122(13):11830–11895
Song JJ, Wei C, Huang ZF et al (2020) A review on fundamentals for designing oxygen evolution electrocatalysts. Chem Soc Rev 49(7):2196–2214
Li Y, Wang TZ, Asim M et al (2022) Manipulating spin polarization of defected Co3O4 for highly efficient electrocatalysis. Trans Tianjin Univ 28(3):163–173
Shinde PV, Samal R, Rout CS (2022) Comparative electrocatalytic oxygen evolution reaction studies of spinel NiFe2O4 and its nanocarbon hybrids. Trans Tianjin Univ 28(1):80–88
Liu F, Shi CX, Guo XL et al (2022) Rational design of better hydrogen evolution electrocatalysts for water splitting: a review. Adv Sci (Weinh) 9(18):e2200307
Chatenet M, Pollet BG, Dekel DR et al (2022) Water electrolysis: from textbook knowledge to the latest scientific strategies and industrial developments. Chem Soc Rev 51(11):4583–4762
Chen FY, Wu ZY, Adler Z et al (2021) Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5(7):1704–1731
Wang XP, Zhong HY, Xi SB et al (2022) Understanding of oxygen redox in the oxygen evolution reaction. Adv Mater 34(50):e2107956
Lu M, Zheng Y, Hu Y et al (2022) Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites. Sci Adv 8(30):eabq3563
Yin J, Jin J, Lu M et al (2020) Iridium single atoms coupling with oxygen vacancies boosts oxygen evolution reaction in acid media. J Am Chem Soc 142(43):18378–18386
Chong LN, Gao GP, Wen JG et al (2023) La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380(6645):609–616
Wang XP, Xi SB, Huang PR et al (2022) Pivotal role of reversible NiO6 geometric conversion in oxygen evolution. Nature 611(7937):702–708
Chen ZC, Guo L, Pan L et al (2022) Advances in oxygen evolution electrocatalysts for proton exchange membrane water electrolyzers. Adv Energy Mater 12(14):2103670
Zhang RR, Guo BB, Pan L et al (2023) Metal-oxoacid-mediated oxyhydroxide with proton acceptor to break adsorption energy scaling relation for efficient oxygen evolution. J Energy Chem 80:594–602
Zhao ZJ, Liu SH, Zha SJ et al (2019) Theory-guided design of catalytic materials using scaling relationships and reactivity descriptors. Nat Rev Mater 4(12):792–804
Grimaud A, Diaz-Morales O, Han BH et al (2017) Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat Chem 9(5):457–465
Huang ZF, Song JJ, Du YH et al (2019) Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts. Nat Energy 4(4):329–338
Wang C, Zhai PL, Xia MY et al (2021) Engineering lattice oxygen activation of iridium clusters stabilized on amorphous bimetal borides array for oxygen evolution reaction. Angew Chem Int Ed 60(52):27126–27134
Wu C, Wang XP, Tang Y et al (2023) Origin of surface reconstruction in lattice oxygen oxidation mechanism based-transition metal oxides: a spontaneous chemical process. Angew Chem Int Ed 62(21):e202218599
Lin YC, Dong Y, Wang XZ et al (2018) Electrocatalysts for the oxygen evolution reaction in acidic media. Adv Mater 35(1):e2210565
Geiger S, Kasian O, Ledendecker M et al (2018) The stability number as a metric for electrocatalyst stability benchmarking. Nat Catal 1(7):508–515
Huang ZF, Xi SB, Song JJ et al (2021) Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst. Nat Commun 12(1):3992
Wang Y, Yang R, Ding YJ et al (2023) Unraveling oxygen vacancy site mechanism of Rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat Commun 14(1):1412
Yao N, Jia HN, Zhu J et al (2023) Atomically dispersed Ru oxide catalyst with lattice oxygen participation for efficient acidic water oxidation. Chem 9:1–15
He ZY, Zhang J, Gong ZH et al (2022) Activating lattice oxygen in NiFe-based (oxy)hydroxide for water electrolysis. Nat Commun 13(1):2191
Wu TZ, Ren X, Sun YM et al (2021) Spin pinning effect to reconstructed oxyhydroxide layer on ferromagnetic oxides for enhanced water oxidation. Nat Commun 12(1):3634
Garcés-Pineda FA, Blasco-Ahicart M, Nieto-Castro D et al (2019) Direct magnetic enhancement of electrocatalytic water oxidation in alkaline media. Nat Energy 4(6):519–525
Zhang RR, Pan L, Guo BB et al (2023) Tracking the role of defect types in Co3O4 structural evolution and active motifs during oxygen evolution reaction. J Am Chem Soc 145(4):2271–2281
Ren X, Wu TZ, Gong ZZ et al (2023) The origin of magnetization-caused increment in water oxidation. Nat Commun 14(1):2482
Li CF, Zhao JW, Xie LJ et al (2021) Surface-adsorbed carboxylate ligands on layered double hydroxides/metal–organic frameworks promote the electrocatalytic oxygen evolution reaction. Angew Chem Int Ed 60(33):18129–18137
Yuan S, Peng JY, Cai B et al (2022) Tunable metal hydroxide–organic frameworks for catalysing oxygen evolution. Nat Mater 21(6):673–680
Huang WZ, Li JT, Liao XB et al (2022) Ligand modulation of active sites to promote electrocatalytic oxygen evolution. Adv Mater 34(18):2200270
Acknowledgements
The authors appreciate the support from the National Key R&D Program of China (2020YFA0710000), the National Natural Science Foundation of China (Nos. 22008170, 22278307, 22222808, 21978200), the Haihe Laboratory of Sustainable Chemical Transformations and the Tianjin Research Innovation Project for Postgraduate Students (2022BKYZ035).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, X., He, Z., Ajmal, M. et al. Recent Advances in the Comprehension and Regulation of Lattice Oxygen Oxidation Mechanism in Oxygen Evolution Reaction. Trans. Tianjin Univ. 29, 247–253 (2023). https://doi.org/10.1007/s12209-023-00364-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12209-023-00364-z