
Abstract
A new scheme for the preparation of highly dispersed precious metal catalysts is proposed in this work. Samples of LaCo1−xPtxO3/SiO2 (x = 0.03, 0.05, 0.07, 0.09, and 0.10) were prepared through a simple method of citrate acid complexation combined with impregnation. In a nanocrystallite of LaCo1−xPtxO3, ions of lanthanum, cobalt, and platinum are evenly mixed at the atomic level and confined within the nanocrystallite. In the reduction process, platinum ions were reduced and migrated onto the surface of the nanocrystallite, and the platinum should be highly dispersed owing to the even mixing of the platinum ions in the precursor. When x = 0.05 or lower, the highest dispersion of Pt could be achieved. The highly dispersed Pt is stable, because of the strong interaction between Pt atoms and the support. The catalysts were characterized by BET surface area, temperature-programmed reduction, X-ray diffraction, transmission electron microscopy, CO temperature-programmed desorption, and turnover frequency. Compared with general precious metal Pt catalysts, the LaCo0.95Pt0.05O3/SiO2 catalyst exhibited better activity for CO oxidation, and it maintained stability at a high temperature of 400 °C for 250 h with complete CO conversion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Supported precious metal catalysts are widely used in the industry for their very good catalytic performance. The size of metal nanoparticles (NPs) is a key element in terms of the catalytic activity. For general metal catalysts, only part of the metal atoms or ions serves as the active sites to play the catalytic role during the reaction. To improve the utilization of noble metals, higher distributions of the metal active sites are expected [1, 2]. Recently, theoretical and experimental results have proved that highly dispersed catalysts have better catalytic activity and selectivity than common metal particles [3]. Particularly for precious metal catalysts, the highly dispersed catalysts can improve the atom catalytic efficiency substantially, thus reducing the cost. At the same time, the highly dispersed metallic atoms make it possible to study the mechanism of heterogeneous catalysis at the molecular level [4, 5].
As mentioned above, highly dispersed catalysts have many advantages, but their preparation is a challenge. The key problem is the high surface free energy of atoms, which promotes aggregation to form small clusters [6]. The reported preparation methods for highly dispersed catalysts include the following: Ramos-Fernandez et al. [7] used the metal organic framework to prepare PTA-MOF catalysts for CO oxidation and the hydrogenation of toluene. Watanabe et al. [8] prepared highly dispersed Pt + Ru alloy clusters through the co-deposition of platinum and ruthenium oxides at the atomic level for the electro-oxidation of methanol. In 2011, Qiao et al. [9] prepared a catalyst of single atom Pt/FeOx by a co-precipitation method for CO oxidation. A photochemical method was used to synthesize Pd/TiO2 monatomic catalysts for styrene hydrogenation [10]. High-temperature heat transfer atom trapping on polyhedral ceria and nanorods was adopted to synthesize thermally stable single-atom platinum-on-ceria catalysts for CO oxidation [11]. The methods above can only be applied to a narrow range, as they are expensive and of low-yield. Therefore, simple and feasible methods for the preparation are needed.
Perovskite-type oxides (PTOs) are mixed oxides with a cubic lattice structure of general formula ABO3, where rare earth elements are usually at the A-site positions and the smaller transition metals are located at the B-sites. The large range of tolerance factors (0.75 ≤ t ≤ 1.0) makes it possible to tolerate significant partial substitution. At the same time, oxygen vacancies caused by doping of perovskites with different species are helpful to the activity of many oxidation reactions. PTOs also have other advantages, such as lower cost and higher thermal stability than noble metals. Furthermore, they exhibit excellent partial substitution tolerance for precious metals (Pd, Rh, and Pt) and are capable of recycling the noble metal atoms in and out of the perovskite lattice in oxidizing and reducing conditions, which can prevent the aggregation of the metal atoms [12,13,14]. The specific surface area of a PTO is low, generally below 10 m2/g [15]. Loading a PTO on a support with high specific surface area is one strategy to overcome this problem.
In this article, a new scheme for preparing highly dispersed catalyst was proposed, as shown in Scheme 1. Taking advantage of the strong interaction between a PTO and precious metals (Pt, here), by supporting a PTO with high specific surface area on silica, it is possible to overcome their disadvantage of low specific surface area. A PTO of LaCo0.95Pt0.05O3 was highly dispersed on SiO2, where the ions of Pt should be uniformly dispersed in the PTO lattice. In the reduction process, ions of Pt should be reduced and migrate onto the surface of the PTO to achieve high dispersions. The as-prepared catalyst showed high activity and very good stability to CO oxidation.

A schematic illustration for preparing the highly dispersed catalyst of Pt/LaCoO3/SiO2
Experimental
Catalyst Preparation
Materials
The catalysts were prepared by a citric acid complexation, combined with an incipient wetness impregnation method. The chemicals involved in the preparation of the catalysts were as follows: La(NO3)3·6H2O, Co(NO3)3·6H2O, and citric acid (C6H8O7) supplied by Aladdin Industrial Corporation, China; H2PtCl6·6H2O procured from Tianjin Delan Fine Chemical Plant; C2H6O2 purchased from Tianjin Guangfu Fine Chemical Research Institute; and silica (SiO2) commercially available from Qingdao Bangkai Separation Material Co., Ltd.
Preparation of LaCo1−xPtxO3/SiO2
LaCo0.95Pt0.05O3/SiO2 is given as the example to illustrate the preparation of LaCo1−xPtxO3/SiO2. Typically, 2 g of LaCo0.95Pt0.05O3/SiO2 was prepared according to the following method. The support, 1.74 g of SiO2, was calcined at 700 °C for 6 h to break down impurities and then dried at 120 °C for 12 h to remove absorbed air and water. The SiO2 support was incipient-wetness-impregnated with an aqueous solution of lanthanum nitrate (1.03 mmol), cobalt nitrate (0.98 mmol), and chloroplatinic acid (0.05 mmol) at a La/Co/Pt molar ratio of 1:0.95:0.05, while citric acid in 120 mol% of the total cations (2.47 mmol) and ethylene glycol in 20 mol% of the citric acid (0.49 mmol) were added. The mixture was left for 24 h to disperse evenly. After complexing for 6 h at 80 °C, the sample was dried for 12 h at 120 °C. Then, the sample was calcined at 350 °C for 2 h with a heating rate of 2 °C/min to decompose citric acid and subsequently was washed to remove chloride with deionized water. After drying at 120 °C for 12 h, the sample was calcined at 700 °C for 6 h with a heating rate of 2 °C/min. The mass fractions of Pt in the calcined catalysts of LaCo0.97Pt0.03O3/SiO2, LaCo0.95Pt0.05O3/SiO2, LaCo0.93Pt0.07O3/SiO2, LaCo0.91Pt0.09O3/SiO2, and LaCo0.90Pt0.10O3/SiO2 are 0.3, 0.5, 0.7, 0.88, and 1%, respectively, and the mass fraction of LaCo1−xPtxO3 in all of the catalysts is 13%.
Similarly, Pt/SiO2 and LaCoPtOx/SiO2 were prepared by the same method with a mass fraction of 0.5% for Pt, except that no citric acid or ethylene glycol was added during the preparation process. LaCoO3/SiO2 was also prepared by the same method with a mass fraction of 13% for the perovskite.
For comparison, unsupported PTOs of LaCo1−xPtxO3 were prepared similarly as for LaCo1−xPtxO3/SiO2, except that no SiO2 support was added for impregnation.
Catalyst Characterization
Specific surface areas and hole structures were calculated by the BET method and the BJH model. The data came from a Micromeritics apparatus, model ASAP-2020, and were collected at −196 °C. Before the measurements, 200 mg of samples were pretreated for 2 h at 300 °C to remove absorbed air and moisture.
H2-temperature-programmed reduction (H2-TPR) experiments were performed on a Thermo-Finnigan TP-5079 which was equipped with a thermal conductivity detector. Samples of 150 mg were heated from 30 to 900 °C at a heating rate of 10 °C/min under a reduction gas mixture of 5 vol% H2/Ar with a flow rate of 25 cm3/min. It should be noted that 19.5 mg of LaCoO3 and 130.5 mg of quartz sand were mixed for the TPR tests.
X-ray diffraction (XRD) experiments were conducted on a Bruker D8-Focus X-ray diffractometer with nickel-filtered Cu Kα radiation (λ = 1.5406 Å). The data range was from 15 to 85° (2θ range), with a scanning speed of 8°/min.
Transmission electron microscopy (TEM) measurements were performed on a Technai G2 F20 transmission electron microscope. The Pt-containing samples were pretreated at 220 °C in hydrogen for 2 h, followed by grinding and dispersing in ethanol. The samples were loaded onto Cu grid for testing.
During CO temperature-programmed desorption (CO-TPD) measurements, the LaCo1−xPtxO3/SiO2 catalysts were first reduced in H2 at 220 °C for 2 h. The chemisorption of CO onto the catalysts was performed in a reactor at atmospheric pressure and 25 °C. The catalyst was outgassed and purged with helium of ultrahigh purity grade (99.9999%, 30 cm3/min) at 400 °C for 0.5 h. The sample was purged at 50 °C for 3 h and allowed to adsorb CO under CO atmosphere for 2 h. Then, TPD measurements were completed at a constant heating rate (5 °C/min) from 50 to 500 °C under helium flow (30 cm3/min) by an SRS RGA300 quadrupole mass spectrometer. The Pt dispersion (D) was calculated based on the following equation [16]:
where MCO is the amount of CO adsorption in μmol; SF is the stoichiometric factor, i.e., the Pt/CO ratio in chemisorption, which is 1 in this study [17]; FWm is the formula weight of the metal Pt; WFm is the weight fraction of the metal Pt in the catalyst; and mcat is the mass of the catalyst.
Based on the Pt dispersion in the catalyst, the turnover frequency (TOF) value at 145 °C was calculated according to the following equation [18]:
where XCO is the CO conversion at 145 °C; FCO is the flow rate of CO in mol/s; mcat is the mass of catalyst; XPt is the Pt loading in the catalyst; DPt is the dispersion of Pt which was calculated based on the CO-TPD results; and MPt is the molar weight of Pt (195.1 g/mol).
Catalytic Performance Test for CO Oxidation
The catalytic activity measurements were performed on a fixed bed micro-reactor at atmospheric pressure. Firstly, a 100 mg sample was reduced at 220 °C for 2 h in H2 atmosphere with a heating rate of 5 °C/min. The feed gases contained 1% CO, 1% O2, and 98 vol% N2 and reached the flow rate of 40 mL/min in total. The catalyst sample was well distributed from 40 to 60 mesh. A thermocouple was inserted into the catalyst bed and used to measure the reaction temperature. The reaction exhaust was analyzed by an online gas chromatograph, SP-3420, which was equipped with a TCD and a 5 A molecular sieve column. The performance of samples was determined by CO conversion, which was defined as follows [19]:
where [CO]in is the concentration of CO in the feed gases and [CO]out is the concentration of CO in the effluent stream.
Results and Discussion
Catalyst Characterizations
N2 Adsorption and Desorption Isotherms
Nitrogen adsorption–desorption isotherms and the BJH pore size distribution of SiO2 and LaCo0.95Pt0.05O3/SiO2 are shown in Fig. 1. The isotherms belong to Type IV under IUPAC classifications [20], which closely relates to the properties of a mesoporous structure. As shown in Fig. 1, the most probable size and the specific surface area declined slightly from 9.45 nm and 369.8 m2/g for the SiO2 support to 8.76 nm and 326.6 m2/g for LaCo0.95Pt0.05O3/SiO2, respectively, which is likely caused by the blockage of some mesopores by the NPs of the PTO. The average pore size (12 nm) is larger than the size of the PTO (below 9 nm), which can be seen in Fig. 4a–c, meaning that the NPs of the PTO can enter the mesopores. The specific surface area of the SiO2 support is large enough to highly disperse the PTO of LaCo0.95Pt0.05O3 when the amount of the PTO loaded is 13 wt%.

Nitrogen adsorption–desorption isotherms and BJH pore diameter distributions, (square) SiO2 calcined at 700 °C, (circle) LaCo0.95Pt0.05O3/SiO2 calcined at 700 °C
Temperature-Programmed Reduction (TPR)
The TPR profiles are shown in Fig. 2. The reduction curves of LaCoO3 have been well studied, where the peak at the low temperature of approximately 400 °C is attributed to the reduction of Co3+ to Co2+, and the peak at high temperature of approximately 570 °C is attributed to the reduction of Co2+ to metallic Co0 [21, 22]. Compared with LaCoO3, for LaCoO3/SiO2, the peak at around 570 °C moved toward a higher temperature, which is due to the effect of the carrier. Compared with LaCoO3/SiO2, the TPR peaks for LaCo1−xPtxO3/SiO2 shifted back to lower temperatures, and the magnitude of the shift increased with the increase in Pt content in the catalyst, which agrees with the shift due to the hydrogen spillover from the sites of Pt as pointed out in previous works [23, 24]. The most important information is that the outlines of the TPR profiles of LaCo1−xPtxO3/SiO2 are similar to that of the PTO of LaCoO3, indicating that PTO was formed as LaCo1−xPtxO3/SiO2.

TPR profiles of a LaCoO3, b LaCoO3/SiO2, c LaCo0.97Pt0.03O3/SiO2, d LaCo0.95Pt0.05O3/SiO2, e LaCo0.93Pt0.07O3/SiO2, f LaCo0.91Pt0.09O3/SiO2, g LaCo0.90Pt0.10O3/SiO2
Ions of Pt in the PTO lattice of LaCo1−xPtxO3/SiO2 can easily be reduced at a reduction temperature generally lower than 300 °C [3, 17, 19]. Additionally, the content of Pt in LaCo1−xPtxO3/SiO2 is small; therefore, no reduction peaks corresponding to the ions of Pt were detected. The TPR results indicate that after reduction, LaCo1−xPtxO3/SiO2 was converted to Pt/LaCoO3/SiO2, which is further confirmed by the following XRD and TEM results.
XRD Results
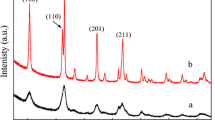
Figure 3A shows XRD patterns of the unsupported PTOs of LaCo1−xPtxO3. The characteristic diffraction peaks of the perovskite phase at 2θ = 32.9, 33.3, 47.5, and 59.0° are clearly seen, and no peaks corresponding to other impurity phases could be observed, suggesting that the PTOs were synthesized well. From the enlarged pattern of LaCo1−xPtxO3 in Fig. 3B, it is seen that the diffraction peaks of the perovskite phase shifted to smaller values of 2θ, which means that the crystal lattice spacing of the perovskite phase increased. The shift increased with the increase in the Pt content in the PTOs. This should be indicative of the imbedding of Pt ions into the lattice of PTO. The size of the Pt ion is larger than the size of the Co ion, leading to the increase in the lattice spacing. Since no diffraction peak corresponding to platinum or platinum oxide was detected, entering of Pt ions into the lattice of PTO is supported.

XRD patterns of A: a LaCoO3, b LaCo0.95Pt0.05O3, c LaCo0.93Pt0.07O3, d LaCo0.91Pt0.09O3, and e LaCo0.89Pt0.11O3; B: the partially enlarged drawing of A; C: a SiO2, b 13% LaCo0.95Pt0.05O3/SiO2, c 25% LaCo0.95Pt0.05O3/SiO2, d 30% LaCo0.95Pt0.05O3/SiO2, e 40% LaCo0.95Pt0.05O3/SiO2, f 50% LaCo0.95Pt0.05O3/SiO2
The XRD patterns of the supported catalysts of LaCo0.95Pt0.05O3/SiO2 and SiO2 are shown in Fig. 3C. Besides the broad diffraction peaks corresponding to SiO2, weak diffraction peaks corresponding to the perovskite phase could be detected, indicating that the PTOs were successfully supported on silica. The peak intensity of PTO increased with the increase in PTO loading amount, and when the loading amount of PTOs is 13 wt%, diffraction peaks of perovskite could hardly be detected, suggesting that the PTOs were highly dispersed, which is consistent with the TEM results in Fig. 4.

TEM images of a fresh LaCo0.95Pt0.05O3/SiO2; b the enlarged view of a in the red circle; c LaCo0.95Pt0.05O3/SiO2 reduced at 220 °C; d the enlarged view of c in the red circle; e LaCo0.90Pt0.10O3/SiO2 reduced at 220 °C; f the enlarged view of e in the red circle
TEM Images
Figure 4a, b shows representative TEM images of the calcined LaCo0.95Pt0.05O3/SiO2. From the images of LaCo0.95Pt0.05O3/SiO2, many of PTO NPs with sizes below 9 nm can be found on the surface of SiO2. Figure 4b shows a representative crystallite of the PTO, with a crystal spacing of 2.74 Å, corresponding to the crystalline plane [110] of LaCo0.95Pt0.05O3.
After reduction at 220 °C, the NPs with an interplanar crystal spacing of 2.72 Å, consistent with crystal spacing of plane [110] for LaCoO3, can be found in Fig. 4d. The slight decrease in the lattice spacing from 2.74 Å in Fig. 4b to 2.72 Å here would be attributed to platinum ions’ escaping from the perovskite lattice. At the same time, no platinum lattices can be detected for LaCo0.95Pt0.05O3/SiO2, indicating that platinum is highly dispersed on the support. It agrees well with XRD results and our assumption in Scheme 1.
The representative images of LaCo0.90Pt0.10O3/SiO2 (Pt loading is 1 wt%) reduced at 220 °C are shown in Fig. 4e, f; the crystallites of LaCoO3 can be seen. The image in Fig. 4f shows a typical metallic Pt NP. The crystal lattice spacing is 2.27 Å, which is in accordance with the crystalline plane [111] of platinum metal. The result indicates that too much Pt added resulted in the comparatively poor dispersion of Pt atoms.
Catalytic Performance
To study the catalytic performance of the supported Pt catalysts, CO oxidation was chosen as the probe reaction, which is an extensively used probe and a very important reaction in environmental protection [25]. CO conversions over the catalysts with the same platinum content of 0.5 wt% are shown in Fig. 5a. The complete CO oxidation temperatures are 240, 260, and 320 °C for LaCo0.95Pt0.05O3/SiO2, LaCoPtOx/SiO2, and Pt/SiO2, respectively. Meanwhile, the highest CO conversion is about 90% for LaCoO3/SiO2 at a temperature of 480 °C. It indicates that the catalyst prepared according to Scheme 1 is highly active.

Variation of CO conversion with reaction temperature for a supported catalysts with the same platinum content of 0.5 wt% or the same PTO content of 13 wt%, b the catalysts of PTO supported on SiO2 with different platinum contents of 0.3, 0.5, 0.7, 0.88%, and 1 wt%. Reaction conditions: 1% CO, 1% O2, 98 vol% N2 balance, space velocity = 24,000 mL/(gcath)
Considering the similar synthesis process and perovskite content between LaCo0.95Pt0.05O3/SiO2 and LaCoO3/SiO2, the huge difference in activity comes from the small amount of metallic platinum. Reducing LaCo1−xPtxO3/SiO2 resulted in Pt/LaCoO3/SiO2, as stated in the discussion about TPR results, over which CO is adsorbed on the highly dispersed platinum and would react with the oxygen species adsorbed in the perovskite vacancies [26]. The mechanism on LaCoO3/SiO2 is generally proposed that oxygen species, adsorbed at the oxygen vacancies on LaCoO3, react with CO adsorbed on Co3+ adjacent to the oxygen vacancies to generate CO2 [26]. Platinum atoms are much more active than Co3+, therefore leading to the much better activity of LaCo1−xPtxO3/SiO2. For Pt/SiO2, CO adsorbed on Pt atoms reacts with oxygen activated on the Pt surface [27], and the platinum species loaded with the regular impregnation method at a calcination temperature of 700 °C is not highly dispersed, thus showing obviously inferior activity. As for LaCoPtOx/SiO2, a PTO was not formed, which means that Pt is not in close contact with the oxide of cobalt, and the dispersion of Pt atoms would not be very high; thus, CO oxidation proceeds via a similar mechanism to LaCo0.95Pt0.05O3/SiO2. Therefore, LaCoPtOx/SiO2 showed inferior activity compared to LaCo0.95Pt0.05O3/SiO2.
Figure 5b shows the variations of CO conversion over the series of catalysts of LaCo1−xPtxO3/SiO2 with the reaction temperature. The complete CO conversion temperatures are 280, 240, 230, 215, and 205 °C over the catalysts, when x is 0.03, 0.05, 0.07, 0.09, and 0.10, respectively, which increases with the increase in the amount of platinum present. This is in the expectation, as a higher content of platinum would provide more active sites. The TOFs, CO converted on per atom of platinum per second, decreased slightly with the increase in platinum content as listed in Table 1, suggesting that elevating the dispersion could improve the utilization efficiency of the noble metal platinum.
As shown in Fig. 6, LaCo0.95Pt0.05O3/SiO2 shows excellent stability at the high temperature of 400 °C during 240 h testing, as complete CO conversion was maintained from the start to the finish. At the same time, the dispersion of Pt atoms slightly decreased from 88.3 to 85.1% after the stability test, and no Pt NPs could be observed from TEM images.

Stability of LaCo0.95Pt0.05O3/SiO2 reduced at 220 °C. Reaction conditions: 1% CO, 1% O2, 98 vol% N2 balance at 400 °C and a space velocity of 24,000 mL/(gcat h)
It has been reported that under oxidation conditions, Pt, Pd, or Rh atoms on the surface of a PTO could enter into the lattice of the PTO support, thus acting as the lattice ions of the PTO. Under reduction conditions, the Pt/Pd/Rh ions in a PTO would be reduced to metal atoms and migrate out onto the surface of the PTO. The “entering into” and “migrating out” may cycle under certain atmospheres, meaning that Pt/Pd/Rh species may be cycling between the surface and lattice of a PTO [13, 14, 34, 35]. This cycling might function for LaCo0.95Pt0.05O3/SiO2 in the CO oxidation reaction process, considering that the reaction gas mixture contains CO/O2 at a ratio of 1:1. Therefore, the highly dispersed platinum atoms showed excellent sintering resistance ability. At least, the Pt species could interact with the support of LaCoO3, and the interaction would prevent the Pt atoms from moving, therefore showing excellent sintering resistance ability.
Table 2 summarizes representative excellent Pt-based catalysts developed in recent years and compares them with the LaCo0.95Pt0.05O3/SiO2 of this work. It can be seen that LaCo0.95Pt0.05O3/SiO2 exhibited excellent stability, which was tested at an obviously higher temperature and maintained for a much longer time compared to other reactions in the table.
To combine the above results, the process shown in Scheme 1 is analyzed as follows. A PTO of LaCo1−xPtxO3 was highly dispersed on SiO2 with a high surface area, and the platinum ions should be uniformly dispersed in the PTO lattice. In the reduction process, platinum ions should be reduced and migrate out from the lattice onto the surface of LaCoO3. In the lattice of LaCo1−xPtxO3, platinum ions were uniformly dispersed; therefore, in the reduction process, platinum ions should migrate out evenly and highly disperse on the surface of LaCoO3. When the content of platinum in LaCo1−xPtxO3 is low, the platinum atoms (migrated from the lattice of LaCo1−xPtxO3) would be highly dispersed. When x in LaCo0.95Pt0.05O3/SiO2 is 0.05 or below, the characterization results suggest that the content of platinum is low enough, as when x = 0.03 and 0.05, the dispersions of Pt are 92.5 and 88.3%, respectively, and no Pt NPs could be found by using high-resolution TEM. It is accepted that Pt atoms can activate CO and the oxygen vacancies can activate oxygen, and the two activated species therefore bond to generate CO2, leading to the high activity for CO oxidation.
Conclusions
Highly dispersed Pt catalysts can be prepared by using PTOs as the precursor via a simple method of citrate acid complexation combined with impregnation. Specifically, in LaCo0.95Pt0.05O3/SiO2, the PTO of LaCo0.95Pt0.05O3 could be highly dispersed on SiO2 supports, and after reduction, Pt/LaCoO3/SiO2 was generated, where the platinum was highly dispersed, since the platinum ions were uniformly dispersed in the precursor of LaCo0.95Pt0.05O3. It is interesting to note that the highly dispersed Pt exhibited excellent resistance to sintering, being attributed to the confinement of Pt atoms by LaCoO3 on SiO2. Thus, the prepared catalyst showed high activity and excellent stability for CO oxidation.
References
Thomas JM, Saghi Z, Gai PL (2011) Can a single atom serve as the active site in some heterogeneous catalysts? Top Catal 54(10–12):588–594
Ranocchiari M, Lothschütz C, Grolimund D et al (2012) Single-atom active sites on metal-organic frameworks. Proc R Soc A Math Phys Eng Sci 468:1985–1999
Job N, Pereira MFR, Lambert S et al (2006) Highly dispersed platinum catalysts prepared by impregnation of texture-tailored carbon xerogels. J Catal 240(2):160–171
Zhai Y, Pierre D, Si R et al (2010) Alkali-stabilized Pt-OHx species catalyze low-temperature water–gas shift reactions. Science 329:1633–1636
Yang M, Li S, Wang Y et al (2014) Catalytically active Au–O(OH)x-species stabilized by alkali ions on zeolites and mesoporous oxides. Science 346(6216):1498–1501
Uzun A, Ortalan V, Browning ND et al (2010) A site-isolated mononuclear iridium complex catalyst supported on MgO: characterization by spectroscopy and aberration-corrected scanning transmission electron microscopy. J Catal 269(2):318–328
Ramos-Fernandez EV, Pieters C, Van der Linden B et al (2012) Highly dispersed platinum in metal organic framework NH2-MIL-101(Al) containing phosphotungstic acid—characterization and catalytic performance. J Catal 289:42–52
Watanabe M, Uchida M, Motoo S (1987) Preparation of highly dispersed Pt + Ru alloy clusters and the activity for the electro-oxidation of methanol. J Electroanal Chem Interfacial Electrochem 229(1–2):395–406
Qiao B, Wang A, Yang X et al (2011) Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat Chem 3:634–641
Liu P, Zhao Y, Qin R et al (2016) Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 352(6287):797–801
Jones J, Xiong H, Delariva AT et al (2016) Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 353(6295):150–154
Katz MB, Zhang S, Duan Y et al (2012) Reversible precipitation/dissolution of precious-metal clusters in perovskite-based catalyst materials: bulk versus surface re-dispersion. J Catal 293:145–148
Jarrige I, Ishii K, Matsumura D et al (2015) Toward optimizing the performance of self-regenerating Pt-based perovskite catalysts. ACS Catal 5(2):1112–1118
Taniguchi M, Tanaka H, Uenishi M et al (2007) The self-regenerative Pd-, Rh-, and Pt-perovskite catalysts. Top Catal 42(1–4):367–371
Kingsley JJ, Pederson LR (1993) Combustion synthesis of perovskite LnCrO3 powders using ammonium dichromate. Mater Lett 18(1–2):89–96
Tanksale A, Beltramini JN, Dumesic JA et al (2008) Effect of Pt and Pd promoter on Ni supported catalysts—a TPR/TPO/TPD and microcalorimetry study. J Catal 258(2):366–377
Lanyon MAH, Trapnell BMW (1955) The interaction of oxygen with clean metal surfaces. Proc R Soc Lond Ser A Math Phys Sci 227(1170):387–399
Jia AP, Jiang SY, Lu JQ et al (2010) Study of catalytic activity at the CuO − CeO2 interface for CO oxidation. J Phys Chem C 114(49):21605–21610
Niu T, Liu GL, Liu Y (2014) Preparation of Ru/graphene-meso-macroporous SiO2 composite and their application to the preferential oxidation of CO in H2-rich gases. Appl Catal B Environ 154–155:82–92
Tang H, Li S, Gong D et al (2017) Bimetallic Ni–Fe catalysts derived from layered double hydroxides for CO methanation from syngas. Front Chem Sci Eng 11(4):613–623
Levasseur B, Kaliaguine S (2008) Methanol oxidation on LaBO3 (B = Co, Mn, Fe) perovskite-type catalysts prepared by reactive grinding. Appl Catal A Gen 343(1–2):29–38
Sun S, Yang L, Pang G et al (2011) Surface properties of Mg doped LaCoO3 particles with large surface areas and their enhanced catalytic activity for CO oxidation. Appl Catal A Gen 401(1–2):199–203
Kozlova EA, Korobkina TP, Vorontsov AV et al (2009) Enhancement of the O2 or H2 photoproduction rate in a Ce3+/Ce4+–TiO2 system by the TiO2 surface and structure modification. Appl Catal A Gen 367(1–2):130–137
Jermwongratanachai T, Jacobs G, Wenping M et al (2013) Fischer–Tropsch synthesis: comparisons between Pt and Ag promoted Co/Al2O3 catalysts for reducibility, local atomic structure, catalytic activity, and oxidation–reduction (OR) cycles. Appl Catal A Gen 464–465:165–180
Birgersson H, Eriksson L, Boutonnet M et al (2004) Thermal gas treatment to regenerate spent automotive three-way exhaust gas catalysts (TWC). Appl Catal B Environ 54(3):193–200
Viswanathan B (1992) CO oxidation and NO reduction on perovskite oxides. Catal Rev 34(4):337–354
Eiswirth M, Möller P, Wetzl K et al (1989) Mechanisms of spatial self- organization in isothermal kinetic oscillations during the catalytic CO oxidation on Pt single crystal surfaces. J Chem Phys 90(1):510–521
Singhania A, Gupta SM (2017) Nanocrystalline ZrO(2) and Pt-doped ZrO(2) catalysts for low-temperature CO oxidation. Beilstein J Nanotechnol 8:264–271
Avgouropoulos G, Ioannides T, Papadopoulou Ch et al (2002) A comparative study of Pt/γ-Al2O3, Au/α-Fe2O3 and CuO–CeO2 catalysts for the selective oxidation of carbon monoxide in excess hydrogen. Catal Today 75:157–167
Li S, Liu G, Lian H et al (2008) Low-temperature CO oxidation over supported Pt catalysts prepared by colloid-deposition method. Catal Commun 9(6):1045–1049
Epling WS, Cheekatamarla PK, Lane AM (2003) Reaction and surface characterization studies of titania-supported Co, Pt and Co/Pt catalysts for the selective oxidation of CO in H2-containing streams. Chem Eng J 93(1):61–68
Roh HS, Potdar HS, Jun KW et al (2004) Low temperature selective CO oxidation in excess of H2 over Pt/Ce—ZrO2 catalysts. Catal Lett 93(3–4):203–207
Xu H, Fu Q, Yao Y et al (2012) Highly active Pt–Fe bicomponent catalysts for CO oxidation in the presence and absence of H2. Energy Environ Sci 5:6313–6320
Tanaka H, Taniguchi M, Uenishi M et al (2006) Self-regenerating Rh- and Pt-based perovskite catalysts for automotive-emissions control. Angew Chem Int Edn 45(36):5998–6002
Tanaka H, Uenishi M, Taniguchi M et al (2006) The intelligent catalyst having the self-regenerative function of Pd, Rh and Pt for automotive emissions control. Catal Today 117(1–3):321–328
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 21576192, 21776214).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fang, C., Zhong, H., Wei, Y. et al. Highly Dispersed Pt Species with Excellent Stability and Catalytic Performance by Reducing a Perovskite-Type Oxide Precursor for CO Oxidation. Trans. Tianjin Univ. 24, 547–554 (2018). https://doi.org/10.1007/s12209-018-0175-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12209-018-0175-1