
Abstract
8p11 myeloproliferative syndrome is a rare hematological malignancy caused by the translocation of FGFR1. Patients present with a myeloproliferative neoplasm that frequently transforms into acute myeloid leukemia or T-lymphoblastic lymphoma/leukemia. Here, we report a molecular study of a patient with 8p11 myeloproliferative syndrome who developed acute B-lymphoblastic leukemia and then transformed to mixed-phenotype acute leukemia. A 67-year-old woman was diagnosed with a myeloproliferative neoplasm with t(6;8)(q27;p12) and was monitored for polycythemia vera. Four years later, she developed acute B-lymphoblastic leukemia with an additional chromosomal abnormality of − 7. Despite two induction regimens, she failed to achieve complete remission, and leukemia transformed into mixed-phenotype leukemia. Targeted sequencing of serial bone marrow samples identified the RUNX1 L144R mutation upon transformation to B-cell leukemia. After those two induction regimens, some RUNX1 mutation-positive leukemic cells obtained the JAK2 V617F mutation, which was associated with the emergence of myeloid markers, including myeloperoxidase.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Eight p11 (8p11) myeloproliferative syndrome (EMS) is a rare hematological malignancy caused by the translocation of FGFR1 located on chromosome 8p11-12. Thirteen partner genes of FGFR1 have been reported [1]. The common fusion genes are ZMYM2-FGFR1 and BCR-FGFR1. The generation of the fusion gene and the resultant expression of the fusion protein leads to constitutive activation of the FGFR1 tyrosine kinase. Patients with EMS present with a myeloproliferative neoplasm (MPN) associated with eosinophilia and lymphadenopathy and frequently progress to acute myeloid leukemia or T-lymphoblastic leukemia/lymphoma or rarely to acute B-lymphoblastic leukemia or mixed-phenotype acute leukemia [, , 2,3,4]. The median time for leukemic transformation is two months [2]. In general, transformed leukemia is refractory to chemotherapy, and thus, hematopoietic stem cell transplantation is the only curative treatment. The mechanism of transformation in EMS is largely unknown.
We present a case with 8p11 syndrome that progressed to acute B-lymphoblastic leukemia, which transformed into mixed phenotype leukemia after two induction regimens. We performed targeted sequencing of serial bone marrow samples to elucidate the molecular mechanisms underlying the development of acute leukemia in MPN.
Case presentation
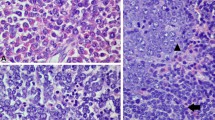
A 67-year-old woman was referred to Dokkyo Medical University Hospital for leukocytosis. Her white blood cell count was 35.3 × 109/L (blast 0%, promyelocyte 0.5%, myelocyte 5.5%, metamyelocyte 6%, neutrophil 69%, eosinophil 3% and basophil 0%), red blood cell count was 6.82 × 1012/L (mean corpuscular volume 73.8 fL, hematocrit 50.3%, and hemoglobin 151 g/L), and platelet count was 230 × 109/L. The serum erythropoietin level was low (1.4 mIU/ml). The bone marrow aspiration showed a hypercellularity with 1.6% blasts (Fig. 1A). G-banding analysis revealed 46, t(6;8)(q27;p12) in 15 cells, 46, idem, add(1)(q32) in one cell, and 46, XX in 4 cells out of 20 metaphases analyzed. Fluorescence in situ hybridization (FISH) analysis revealed a split signal of FGFR1 in 91% of the bone marrow mononuclear cells. A CT scan showed moderate splenomegaly. JAK2 V617F mutation was not detected. She was diagnosed with 8p11 myeloproliferative syndrome and was followed up for polycythemia vera without any treatment.

Images of bone marrow aspirate and flow cytometric analyses. A Low-power field (right) and high-power field (left) of the bone marrow aspirates at the time of polycythemia vera. B The bone marrow smear (left) and surface marker expression (right) at the diagnosis of B-ALL. C The bone marrow smear (left) and surface marker expression (right) at the transformation into mixed-phenotype
Four years after the diagnosis, her white blood cell count increased to 161 × 109/L with 2% blasts, and her hemoglobin level and platelet count dropped to 47 g/L and 75 × 109/L, respectively. The bone marrow was hypercellular with 17.8% peroxidase-negative blasts. Flow cytometric analysis showed that the blasts were positive for CD10, CD19, CD22, CD34, and terminal deoxynucleotidyl transferase (TdT) and partially positive for CD13. In addition to t(6;8)(q27;p12), monosomy 7 was detected. Ruxolitinib treatment resulted in no change in the white blood cell count. However, the white blood cell count dropped transiently following the administration of hydroxyurea. Two months later, the blasts in the peripheral blood had increased to 24%. The bone marrow aspiration showed a hypercellular marrow with 70.4% blasts, which expressed the same surface antigens as that of two months before in addition to CD13 (Fig. 1B). Blasts were myeloperoxidase (MPO)-negative by flow cytometric analysis. G-banding analysis revealed 45, XX, t(6;8)(q27;p11.2), -7 in all 20 of the metaphases analyzed (Fig. 2A). Nested reverse transcription-polymerase chain reaction (RT‒PCR) analysis detected the FGFR1OP-FGFR1 fusion transcript using the FGFR1OP and FGFR1 primers as previously described [, 5, 6]. Sequencing of the PCR product indicated that exon 7 of FGFR1OP was fused to exon 9 of FGFR1 (Fig. 2B). Based on these findings, the patient was diagnosed with B-ALL associated with FGFR1 rearrangement. She received induction therapy using the L-AdVP regimen with 20 mg/m2/day daunorubicin on Days 1–3, 19, 20, 33, and 34, and 1.4 mg/m2/day vincristine on Days 1, 8, and 33. L-Asparaginase was administered at 4000 U/day from Day 19 to Day 32. Prednisolone was administered at 40 mg/m2/day from Day 1 and was reduced gradually and ceased on Day 27. As the proportion of blasts in the bone marrow was 9.6% after induction therapy, we considered it a failure. She was subsequently treated with a hyper-CVAD regimen at a 20% dose reduction [7], which reduced the percentage of bone marrow blasts (7.8%). Flow cytometric analysis showed that the blasts were positive for CD13, CD34, CD117, and TdT and partially positive for CD19, CD22, CD33, and MPO but negative for CD10 (Fig. 1C). The leukemic phenotype was judged to transform into a mixed phenotype. Two weeks later, blasts in the bone marrow increased to 42%. She was treated with the JALSG ALL-202-O induction regimen but did not achieve complete remission. Then, she received MEC therapy at a 50% dose reduction [8]. Four weeks after MEC therapy, the white blood cell count was 98.1 × 109/L, and the number of blasts in the bone marrow was 40%. After cytoreduction by prednisolone and cytarabine, she started blinatumomab therapy. Eighteen days after the administration of blinatumomab, as the white blood cell count increased to 97.6 × 109/L, hydroxyurea was added. After the first course of blinatumomab, blasts in the bone marrow were 5.8%. The best supportive care was provided due to poor performance status afterward. She died 11 months after the diagnosis of B-ALL.

Sequencing and cytogenetic analyses of bone marrow cells at the time of diagnosis of B-ALL. A G-banding analysis: the arrows indicate t(6;8)(q27;p11.2). B Sequencing analysis of the reverse transcription-polymerase chain reaction product. The vertical line indicates the junction of FGFR1 and FGFR1OP
We performed targeted-capture sequencing of the patient’s bone marrow cells at the time of the initial increase in blast count, the diagnosis of B-ALL, after the hyper-CVAD regimen, before the administration of blinatumomab and during the administration of prednisolone for a panel of common driver genes implicated in myeloid malignancies [9]. The ALL panel was also used for the sample collected at the time of diagnosis of B-ALL. The analysis was approved by the Institutional Review Boards for Clinical Research of Dokkyo Medical University and Kyoto University in accordance with the Declaration of Helsinki. Written informed consent from the patient was obtained. The clinical course of the patient is summarized in Fig. 3. Although no known driver mutations were identified at the initial increase in blast count, RUNX1 L144R mutation was identified at a variant allele frequency (VAF) of 48% at the time of diagnosis with B-ALL. After the hyper-CVAD regimen, the JAK2 V617F mutation emerged at a VAF of 12% in addition to the RUNX1 L144R mutation at a VAF of 43%. Before the administration of blinatumomab, while the RUNX1 L144R mutation existed at a VAF of 39%, the JAK2 mutation disappeared.

Clinical course and results of cytogenetic and targeted-capture sequencing analyses of bone marrow cells. MEC mitoxantrone, etoposide, cytarabine; VAF variant allele frequency
Discussion
This patient was initially diagnosed with MPN with t(6;8)(q27;p12) and was followed up for polycythemia vera. Four years later, she developed acute B-lymphoblastic leukemia with an additional chromosomal abnormality of -7. After two induction regimens, she failed to achieve complete remission, and leukemia transformed into mixed-phenotype leukemia.
EMS was categorized as myeloid/lymphoid neoplasms with FGFR1 rearrangement in the 2017 revision of the World Health Organization classification of myeloid neoplasms and acute leukemia [10]. To date, 13 translocation partners have been reported [1]. The most common translocation is t(8;13)(p11.2;q12), which generates the ZMYM2-FGFR1 fusion gene [2]. Patients show eosinophilia and often develop lymphadenopathy and T-lymphoblastic lymphoma. The t(8;22)(p11;q11) generating BCR-FGFR1 is another common translocation in patients with EMS. Patients with t(8;22)(p11;q11) present with basophilia rather than eosinophilia, which is similar to chronic myeloid leukemia. Most patients with BCR-FGFR1 develop B-ALL or lymphoma [11]. The prognosis of patients with EMS is poor. The 1-year overall survival is reported to be 43% [12]. Hematopoietic stem cell transplantation is the only curative treatment. The t(6;8)(q27;p11.2)-generating FGFR1OP-FGFR1 is a rare type of EMS. Of note, patients with t(6;8)(q27;p11.2) often present with polycythemia vera, as seen in this case [5, 6]. In our case, the FGFR1OP-FGFR1 fusion transcript was generated between exon 9 of FGFR1 and exon 7 of FGFR1OP, as previously reported [6]. Exon 5 or 9 of FGFR1OP can also fuse with FGFR1 [6, 13]. To date, 12 cases with t(6;8)(q27;p11.2) have been reported, and six patients transformed to acute myeloid leukemia [5, 13,14,15] and one patient developed B-ALL [6] (Table 1). Our case transformed to B-ALL associated with monosomy 7. Notably, the case that Vizmanos reported also carried monosomy 7 at the development of B-ALL [6]. Finally, leukemic cells in our case obtained myeloid markers including MPO in addition to the lymphoid markers after a few courses of chemotherapy for ALL.
In our case, the accumulation of chromosomal abnormalities and genetic mutations was observed during the progression of the disease. At the initial increase in blast count, monosomy 7 was found in all of the leukemic cells and no other gene mutations were observed. After the administration of ruxolitinib and hydroxyurea, she was diagnosed with B-ALL and leukemic cells with RUNX1 mutation expanded at the development of B-ALL. This expansion is probably due to clonal selection associated with those treatments. RUNX1 mutations were frequently observed in patients with EMS, and all patients with RUNX1 mutation presented with acute leukemia [17]. Therefore, RUNX1 mutation may play an important role in the transformation of EMS to leukemia. In our case, the JAK2 V617F mutation emerged after the hyper-CVAD regimen. At the same time, blasts expressed MPO as well as myeloid surface markers. Considering that the JAK2 V617F mutation is a characteristic gene mutation of myeloproliferative neoplasms [18,19,20,21], the emergence of the JAK2 V617F mutation might be associated with the appearance of a myeloid phenotype.
As with chronic myeloid leukemia (CML), constitutive activation of tyrosine kinase in hematopoietic stem cells leads to the development of MPN in EMS [2], and the accumulation of gene mutations causes a blast crisis. For CML, tyrosine kinase inhibitors are effective [22,23,24]. A deep molecular response can be achieved, and progression to blast crisis is rare. For EMS, ponatinib in combination with chemotherapy was reported to be effective for a patient with t(8;22)(p11;q11) as a bridge to hematopoietic stem cell transplantation [4]. In addition, pemigatinib, which is a selective FGFR1-3 inhibitor, is reported to be a promising drug. The phase 2 study of pemigatinib showed that 75% of patients with EMS achieved complete cytogenetic response [25]. Currently, the prognosis of patients with EMS who progress to leukemia is poor, and hematopoietic stem cell transplantation should be performed for transplant-eligible patients as soon as possible. Information on the accumulation of genetic mutation is helpful in selecting an optimal transplant timing. We expect that next-generation sequencing-based multigene panel testing in hematological malignancies will be introduced to clinical practice shortly.
Data availability
The data of this study are available on request from the corresponding author. The data are not publicly available due to privacy.
References
Vega F, Medeiros LJ, Bueso-Ramos CE, Arboleda P, Miranda RN. Hematolymphoid neoplasms associated with rearrangements of PDGFRA, PDGFRB, and FGFR1. Am J Clin Pathol. 2015;144:377–92.
Jackson CC, Medeiros LJ, Miranda RN. 8p11 myeloproliferative syndrome: a review. Hum Pathol. 2010;41:461–76.
Haslam K, Langabeer SE, Kelly J, Coen N, O’Connell NM, Conneally E. Allogeneic hematopoietic stem cell transplantation for a BCR-FGFR1 myeloproliferative neoplasm presenting as acute lymphoblastic leukemia. Case Rep Hematol. 2012;2012: 620967.
Khodadoust MS, Luo B, Medeiros BC, Johnson RC, Ewalt MD, Schalkwyk AS, et al. Clinical activity of ponatinib in a patient with FGFR1-rearranged mixed-phenotype acute leukemia. Leukemia. 2016;30:947–50.
Popovici C, Zhang B, Grégoire MJ, Jonveaux P, Lafage-Pochitaloff M, Birnbaum D, et al. The t(6;8)(q27;p11) translocation in a stem cell myeloproliferative disorder fuses a novel gene, FOP, to fibroblast growth factor receptor 1. Blood. 1999;93:1381–9.
Vizmanos JL, Hernández R, Vidal MJ, Larráyoz MJ, Odero MD, Marín J, et al. Clinical variability of patients with the t(6;8)(q27;p12) and FGFR1OP-FGFR1 fusion: two further cases. Hematol J. 2004;5:534–7.
Kantarjian H, Thomas D, O’Brien S, Cortes J, Giles F, Jeha S, et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer. 2004;101:2788–801.
Amadori S, Arcese W, Isacchi G, Meloni G, Petti MC, Monarca B, et al. Mitoxantrone, etoposide, and intermediate-dose cytarabine: an effective and tolerable regimen for the treatment of refractory acute myeloid leukemia. J Clin Oncol. 1991;9:1210–4.
Ochi Y, Kon A, Sakata T, Nakagawa MM, Nakazawa N, Kakuta M, et al. Combined cohesin-RUNX1 deficiency synergistically perturbs chromatin looping and causes myelodysplastic syndromes. Cancer Discov. 2020;10:836–53.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Montenegro-Garreaud X, Miranda RN, Reynolds A, Tang G, Wang SA, Yabe M, et al. Myeloproliferative neoplasms with t(8;22)(p11.2;q11.2)/ BCR-FGFR1: a meta-analysis of 20 cases shows cytogenetic progression with B-lymphoid blast phase. Hum Pathol. 2017;65:147–56.
Umino K, Fujiwara S-I, Ikeda T, Toda Y, Ito S, Mashima K, et al. Clinical outcomes of myeloid/lymphoid neoplasms with fibroblast growth factor receptor-1 (FGFR1) rearrangement. Hematology. 2018;23:470–7.
Sohal J, Chase A, Mould S, Corcoran M, Oscier D, Iqbal S, et al. Identification of four new translocations involving FGFR1 in myeloid disorders. Genes Chromosomes Cancer. 2001;32:155–63.
Vannier JP, Bizet M, Bastard C, Bernard A, Ducastelle T, Tron P. Simultaneous occurrence of a T-cell lymphoma and a chronic myelogenous leukemia with an unusual karyotype. Leuk Res. 1984;8:647–57.
Chaffanet M, Popovici C, Leroux D, Jacrot M, Adélaïde J, Dastugue N, et al. t(6;8), t(8;9) and t(8;13) translocations associated with stem cell myeloproliferative disorders have close or identical breakpoints in chromosome region 8p11-12. Oncogene. 1998;16:945–9.
Lourenco GJ, Ortega MM, Freitas LLL, Bognone RAV, Fattori A, Lorand-Metze I, et al. The rare t(6;8) (q27;p11) translocation in a case of chronic myeloid neoplasm mimicking polycythemia vera. Leuk Lymphoma. 2008;49:1832–5.
Strati P, Tang G, Duose DY, Mallampati S, Luthra R, Patel KP, et al. Myeloid/lymphoid neoplasms with FGFR1 rearrangement. Leuk Lymphoma. 2018;59:1672–6.
James C, Ugo V, Le Couédic J-P, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8.
Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61.
Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–17.
Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–70.
Saglio G, Kim D-W, Issaragrisil S, le Coutre P, Etienne G, Lobo C, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–9.
Gotlib J, Kiladjian J-J, Vannucchi A, Rambaldi A, Reiter A, Shomali W, et al. A phase 2 study of pemigatinib (FIGHT-203; INCB054828) in patients with myeloid/lymphoid neoplasms (MLNs) with fibroblast growth factor receptor 1 (FGFR1) rearrangement (MLN FGFR1). Blood. 2021;138:385.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Yasuhito Nannya reports consulting fees from Novartis Pharmaceuticals. Motoshi Ichikawa reports honoraria from Novartis Pharmaceuticals. Seishi Ogawa reports grants from Japan Agency for Medical Research and Development (AMED), MEXT, and Japan Society for the Promotion of Science (JSPS), and consulting fees from Novartis Pharmaceuticals. Kinuko Mitani reports grants from Astellas, and honoraria from Astellas, Novartis Pharmaceuticals and Amgen.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Nakamura, F., Seo, S., Nannya, Y. et al. Progression to B acute lymphoblastic leukemia in 8p11 myeloproliferative syndrome with t(6;8)(q27;p12). Int J Hematol 118, 388–393 (2023). https://doi.org/10.1007/s12185-023-03577-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-023-03577-z