
Abstract
Few studies identifying genomic aspects in pediatric acute myeloid leukemia patients in Latin American countries have been reported. The aim of this study was to identify genomic alterations, clinical characteristics and outcomes in a cohort of pediatric AML patients. This descriptive observational cohort study included patients with confirmed de novo acute myeloid leukemia up to 18 years of age. Cytogenetics and conventional FISH analysis, next-generation sequencing and PCR testing were performed. The correlation of genomic data with treatment response and outcomes were analyzed. Of the 51 patients analyzed, 67.4% had a cytogenetic abnormality and 74.5% had a genetic variant. FLT3 variants (ITD or TKD D835) were found in 27.4%, followed by NRAS (21.6%), KRAS (13.7%) and WT1 and KIT (11.8%). Patients were stratified by risk (66.6% high-risk) after the end of induction. FLT3-ITD was associated with relapse (OR 11.25; CI 1.89–66.72, p 0.006) and NRAS with death during induction (OR 16.71; CI 1.51–184.59, p 0.022). Our study highlights the importance of rapid incorporation of genetic testing in pediatric AML in Colombia, as it directly affects treatment decisions and outcomes. Incorporation of targeted therapies with conventional chemotherapy is an increasingly urgent need in pediatric patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous neoplasm representing the second most common type of acute leukemia in pediatric age, with less favorable outcomes than acute lymphoblastic leukemia, reaching overall survival rates that do not exceed 70% in developed countries and relapse rates that vary between 25 and 50% [1, 2], these outcomes are less favorable in developing countries.
Genetic alterations and clinical characteristics in pediatric AML have been studied previously, the majority have been carried out in European and North American population (which includes Hispanics living in the United States), but there is little information about the biological characterization and clinical outcomes in Colombian pediatric patients with de novo AML, even less on the molecular aspects [3]. This lack of knowledge directly affects risk stratification, treatment and survival; hence the importance of conducting this type of studies in our population. The aim of this study was to describe and correlate the genetic alterations, clinical characteristics and outcomes in a cohort of pediatric patients with de novo AML in two pediatric cancer centers from Bogotá, D.C., Colombia.
Methods
Patients, diagnosis, treatment and risk stratification
Descriptive observational cohort study, fifty-one patients between 1 month and 18 years of age with a confirmed diagnosis of de novo acute myeloid leukemia (non-promyelocitic) were included by convenience, with prior informed consent between March 2015, and June 2021, from two main pediatric cancer centers of Colombia, HOMI Fundación Hospital Pediátrico La Misericordia and Clínica Infantil Colsubsidio, Bogotá, D.C., Colombia. Patients with Down syndrome or secondary AML were not included. This protocol was approved by the institutional ethics committee of the Universidad Nacional de Colombia, HOMI Fundación Hospital Pediátrico La Misericordia and Clínica Infantil Colsubsidio. Diagnosis was made on bone marrow aspirate sample, including morphology FAB classification and immunophenotyping by multiparametric flow cytometry (MFC) analysis using EuroFlow Panel. Institutional protocol is based on the national clinical practice guideline [4], all patients received two induction cycles of “7 × 3” with cytarabine 100 mg/m2/d on days 1–7 and daunorubicin 60 mg/m2/d on days 1, 3 and 5. Additionally, all patients received triple intrathecal chemotherapy as prophylaxis of CNS involvement on days 1 and 7. Risk stratification was based on cytogenetic and molecular criteria, along with treatment response (supplemental material).
Post-induction treatment (consolidation) consisted in chemotherapy with 2–3 cycles of intermediate dose of cytarabine 3 gr/m2/day during 3 days plus triple intrathecal chemotherapy in standard-risk patients. For patients within intermediate-risk group, with matched related donor received hematopoietic stem cell transplantation (HSCT) after one or two consolidation cycles and patients without a matched donor received chemotherapy with 2–3 consolidation cycles. Finally, the high-risk group received HSCT with the best available donor after one or two consolidation cycles.
Outcomes and definitions
Response to induction treatment was defined as Complete Remission (CR) with less than 5% of blast by morphology after second induction cycle with complete hematological recovery (≥ 1000/µL leukocytes, ≥ 500/µL neutrophil granulocytes and ≥ 50,000/µL platelets), without blasts in peripheral blood and without extramedullary compromise; CR with incomplete hematologic recovery (iCR) as less than 5% blasts by morphology after the second induction cycle without hematological recovery as described above; Induction Failure (IF) as more that 5% of blasts by morphology in bone marrow after second induction cycle; Resistant Disease as failure to achieve complete remission after first line therapy which includes induction and consolidation (at least two cycles); Treatment Related Mortality was defined as death occurring during treatment and after achieving complete remission, and Transplant Related Mortality as death occurring during transplant process and not related to relapse [5, 6]. Relapse was defined as reappearance of blasts post-CR in peripheral blood, bone marrow or extra bone marrow locations without any other attributable cause. Death during induction was defined as patients dying during induction period or before hematological recovery after end of induction. Measurable residual disease (MRD) was evaluated by flow cytometry with 0.1% as cut-off level after day 21 of second induction cycle. Toxicity was defined according to Common Terminology Criteria for Adverse Events (CTCAE) [7] (supplemental material). Only toxicities grades 3 and 4 were described. Relapse-Free Survival (RFS) was calculated as the time from the first remission to relapse and Overall Survival (OS) was defined as the time from diagnosis to death or last contact alive [8]. For survival analysis, a two-year probability for relapse free survival (RFS) was calculated.
DNA and RNA extraction
DNA was extracted from 200 µL of bone marrow using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's specifications. The DNA purity quantification and verification were performed using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, United States). The DNA obtained was stored at − 20 °C until use. RNA was isolated from bone marrow samples in EDTA using the Quick-RNA MiniPrep Plus kit (ZYMO RESEARCH, Irvine, CA, USA) following the manufacturer's recommendations. The extracted RNA was converted into cDNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, San Francisco, CA, USA) following the manufacturer’s recommendations. The DNA purity quantification and verification were assessed using a NanoDrop™ 2000 (Thermo Fisher Scientific, Waltham, Massachusetts, United States). The RNA was stored at -80 °C, and the cDNA was stored at − 20 °C until use.
Cytogenetics and FISH
Cell culture was performed to obtain metaphases for the chromosomal study with G and Q bands according to standardized protocols [9]. Chromosome visualization was performed using GenASIs (Applied Spectral Imaging, Carlsbad, CA, USA). At least 25 metaphases per sample were analyzed, and the nomenclature was described according to the recommendations of the International System for Human Cytogenomic Nomenclature (ISCN) 2020. FISH was performed to detect the following recurrent rearrangements: t(8;21) (ETO-AML1 [RUNX1-RUNX1T1] Translocation, Dual Fusion Probe, Cytocell, Cambridge, UK) and Inv(16) (CBFB/MYH11 Translocation, Dual Fusion Probe, Cytocell). At least 100 nuclei per study were analyzed, and the interpretation was performed by two independent observers using GenASIs. In some cases, an MLL [KMT2A] Breakapart Probe, Cytocell) was used to confirm cytogenetic findings. Cytogenetic nomenclature was described according to the ISCN 2020 recommendations.
AML Gene panel and rearrangements by NGS
We analyzed 30 genes (ABL1, ASXL1, BRAF, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, HRAS, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NPM1, NRAS, PTPN11, RUNX1, SETBP1, SF3B1, SRSF2, TET2, TP53, U2AF1, WT1, ZRSR2) using the Myeloid Plus kit by SOPHIA Genetics (Sophia Genetics SA, Saint Sulpice, Switzerland) panel by next generation sequencing (NGS) according to manufacturer’s recommendations. In addition, 119 fusions were studied with RNA Myeloid Plus Solution. Sequencing was carried out in an Illumina MiSeq (Illumina, San Diego, CA, USA) and sequencing analysis data with Sophia DDM® software 5.2.7.1 (Sophia Genetics SA, Saint Sulpice, Switzerland). FLT3, NPM1 and CEBPA genes were also analyzed by rapid PCR testing as described previously [10,11,12,13]. Gene variants found by rapid testing in these three genes were confirmed by NGS. Subsequently, each variant was functionally annotated and categorized according to their pathogenicity, following the recommendations international consensus of the Association of Molecular Pathology, American Society of Clinical Oncology and the College of American Pathologists “Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer” (2017) [14].
Statistical analysis
Statistical analysis of quantitative variables is reported as means or medians with dispersion measures given in standard deviation and ranges, according to the nature and distribution of the variables, according to the Shapiro–Wilks normality test to establish the use of parametric tests or non-parametric. Qualitative variables were analyzed with Pearson’s Chi square test and Fisher’s exact test. Statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS) for Windows, version 25.0. A value of p < 0.05 was considered significant. Statistical analyses of quantitative variables were carried out with measures of central tendency and dispersion, means and standard deviations, or medians and ranges, according to their distribution after analysis with normality tests (Kolmogorov–Smirnov or Shapiro–Wilk) to establish the behavior of the data as parametric or non-parametric. Medians were compared using the Mann–Whitney U test for independent non-parametric samples. For bias control, all diagnosed children entered the study, thus minimizing selection bias. Validated techniques were used, as well as positive and negative controls with sample processing. Information was double-checked when entered into the database for quality control.
Results
Clinical characteristics
Fifty-one patients were included. The median age was 10 years (range 0.15–18 years), with an M:F ratio of 1.42:1. Median leukocyte count at diagnosis was 25,580 (1190–1,896,000) and CNS involvement was present in 16 cases (31.37%). Patients were stratified as high risk 34 (66.6%) after the end of induction. During treatment, 96% of patients had at least one toxicity event, the most frequent were mucositis, colitis and transaminitis. Twenty-two out of 33 (66%) patients who received HSCT had a related toxicity event (Table 1).
Cytogenetics and FISH analysis
Five patients had a non-informative karyotype due to absence of metaphases to be analyzed. In the remaining 46 patients, 31 patients (67.4%) had either structural or numeric alterations, 8 (26%) had two or more chromosomal abnormalities (range 2–5). Twenty-four patients (51%) presented gene fusions detected by conventional cytogenetics and FISH. Eleven patients (23%) presented other numerical or structural alterations.
In 15 patients, karyotype or FISH analysis did not show any alteration (32.6%). Among the 31 patients with a cytogenetic alteration, 9 patients had KMT2A (MLL) gene rearrangements (19.6%); 7 patients had an t(8;21) (ETO-AML1 [RUNX1-RUNX1T1] translocation (15.2%); 5 patients (10.9%) had inv(16)(CBFB-MYH11), and 10 patients had other structural or numeric gene rearrangements (21.7%) (Table 1).
AML gene panel by next generation sequencing gene panel
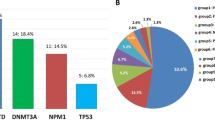
Genetic variants were found either alone or in combination with other genetic variants by NGS in 38/51 patients (74.5%). Genetic variants above variant allele frequency (VAF) 5% were considered clinically relevant. The most frequent pathogenic genetic variant found was in FLT3. Fourteen patients (25.5%) had either ITD (8/14), TKD D835 (5/14) or both ITD/TKD (1/14). The second most frequent genetic alteration was in NRAS (codon 12, 13, 59 or 61) in 11 patients (21.6%), followed by KRAS variants in 7 (13.7%), WT1 and KIT genetic variants in 6 patients, respectively (11.8%), CEBPA (5, 9.8%), U2AF1 (3, 5.9%), NPM1, PTPN11, ASXL1, ETV6 and IDH1 (2, 3.9%), RUNX1, EZH2 and CBL1 (1, 2% each) (Table 1). Regarding WT1 gene mutations, 2 patients had nonsense variants, 2 frameshift mutations and 2 patients had double mutations. It is worth it to mention that no DNMT3A mutations were found in our cohort.
Genomic alterations, co-mutation patterns and clinical outcomes analysis
Eleven out of 12 patients with Core Binding Factor (CBF) alterations had mutations involving signaling pathways (RAS/KIT/FLT3/WT1). All KIT mutations are related to CBF fusions (4 with t(8;21) and 2 with inv(16)). Two out of 4 patients with t(8;21) and KIT mutations died (50%) (death related to HSCT). Patients with Inv(16) and KIT mutations are alive post-HSCT. In addition, 80% of patients with Inv(16) (4/5) carried FLT3 gene variants (3 with TKD and 1 ITD)(2 dead related to HSCT, 1 in relapse, 1 alive in remission) (Tables 1 and 2).
Twelve out of 14 patients (85.7%) with FLT3 mutations were classified as high risk after the end of induction and received HSCT as part of their treatment. Four patients had a high allelic ratio (> 0.5) and 4 had a low allelic ratio. Only 3 out of 14 patients are alive and in remission (21.4%). We found significant association between FLT3-ITD with relapse 11.25 OR (CI 1.89–66.72, p 0.006) and lower blast clearance at day 21 after first induction cycle (3.69% median blast count in bone marrow range 0–55), while FLT3 negative samples had a 1.35% median blast count in bone marrow (range 0–84). However, these differences did not reach a statistically significant difference. Six out of 9 FLT3-ITD patients died post-HSCT. All patients with high allelic ratio in FLT3-ITD that were classified as high risk after the end of induction died (2 due to relapse and 2 due to failed remission). Three out of four patients with low allelic ratio in FLT3-ITD died (2 relapse and 1 HSCT related death). No relationship was found with leukocyte counts or with a specific morphologic subtype. In 10 FLT3 positive patients (71%) age was ≥ 10 years; there were no FLT3 cases in children under 2 years of age, despite representing 25% of the cohort. All patients with WT1 had FLT3 mutations; four out of 6 patients died (66.6%). In addition, NRAS mutations were strongly associated with death during induction 16.71 OR (CI 1.51–184.59, p 0.022). No other significant associations were found in our cohort for other genetic variants (Table 3). Two-year probability for RFS was 25% in our cohort (Fig. 1).

Kaplan–Meier curve for Relapse Free Survival
Discussion
Despite growing data on pediatric AML and its genetic alterations, few studies have been carried out in the Latin American population outside of the US reflecting the clinical and molecular characteristics [15, 16]. Cytogenetic alterations in pediatric AML can be divided broadly into different specific groups: 25% CBF, 20% KMT2A rearrangements, and 20% normal karyotype. These alterations have a particular distribution pattern associated with age [17]. We found similar frequencies in the group of patients reported here with AML, CBF 26.1%, KMT2A 19.6% and normal karyotype 32.6%, respectively. In other countries of the region such as Peru, the frequency of cytogenetic alterations in a pediatric cohort from a reference hospital was recently described; 60.8% of patients had a chromosomal alteration, being t(8;21) the most frequent [15]. A report from Argentina showed similar results, 32% of normal karyotype, 17% of t(8;21), but discrepant for KMT2A 1.8% using only karyotype [16].
More than 90% of pediatric AML cases have at least one molecular alteration; the majority of cases with a normal karyotype [18]. The most frequent gene involved in pediatric AML is FLT3 [10, 17,18,19,20]. A similar result was found in our cohort with 25.5% FLT3 positive patients first by FLT3 PCR and by NGS. However, in a report with more than 1000 AML patients, NRAS mutations were the most frequent mutations (nearly 30%) found in children with AML, followed by FLT3 [18]. In another report, a low frequency of FLT3 involvement in 27 Japanese patients was reported [21], however, the latter results should be taken carefully since NGS was used to characterize FLT3-ITD mutations, and unless the bioinformatic approach used is designed to detect large insertions, as the case of most FLT3-ITD mutations, it might be missed by NGS.
FLT3-ITD is known as an unfavorable prognostic marker [17, 19, 20, 22]. Our results corroborate this finding in Colombian patients, since 12 out of 14 patients (85.7%) were classified as high-risk patients at the end of induction and FLT3-ITD was found as a risk factor for relapse 11.25 OR (CI 1.89–66.72, p 0.006).
As mentioned previously, NRAS mutations was the most frequent mutation found among pediatric AML patients in one of the largest cohorts of AML pediatric patients studied thus far [[18]]. In our cohort, NRAS mutation was the second most frequent genetic alteration found (21.6%) and was strongly associated with death during induction 16.71 OR (CI 1.51–184.59, p 0.022), followed by KRAS (11.7%) and WT1 (11.8%). Thus, FLT3, RAS and WT1 were the most frequent mutations found in our cohort as has been reported earlier for a large cohort in the US [18]. Few studies have evaluated the clinical significance of RAS signaling pathway alterations in pediatric patients with AML. In the Japanese clinical trial AML-05, which includes more than 400 patients, NRAS was the most frequently involved, associated with a favorable prognosis, especially in the presence of CBFB-MYH11 fusion [23]. In our cohort of 11 NRAS positive cases, 4 were also FLT3 positive (3 ITD, one TKD), 2 had CBFB-MYH11 fusion, and one RUNX1-RUNX1T1 fusion. Currently, one NRAS positive patient with co-expression of CBFB-MYH11 fusion and FLT3-TKD is alive and in remission.
Few studies have addressed the prognostic effect of KIT variants in pediatric patients [24]. The prognostic significance of KIT mutations in CBF is still controversial as a potential risk factor for prognosis [24]. In a systematic review, KIT mutations were related to relapse and poor relapse free survival, especially with FLT3-TKD D835 mutations. They were also seen at a higher frequency related to WBC increments, especially in patients with Inv(16) [25]. In a large study from patients between 16 and 64 years of age, a close association was found between adverse effects with KIT mutations and RUNX1-RUNX1T1 but not with CBFB-MYH11 [24]. In our cohort, all patients with RUNX1-RUNX1T1 fusion and KIT mutations were classified as high risk after the end of induction (MRD > 1 after the first cycle of induction), 50% have died.
Co-occurrence of WT1 with FLT3-ITD mutations are frequently associated with induction failure and dismal outcomes in children with AML (p < 0.0001) [2, 18]. In our cohort, we found that 6 patients carrying WT1 mutations were also FLT3 positive. However, this genomic relation did not show any statistically significant association with induction failure, likely due to the sample size of our study.
Despite treatment intensification with HSCT in patients with high-risk disease due to cytogenetic/molecular characteristics and poor response to induction therapy, outcomes continue to be unfavorable. In our cohort, 65% (33/51) of our patients had an indication for HSCT, currently 15 out of 33 (45%) are alive and in remission. However, in patients with FLT3-ITD mutations, this proportion was lower, 3 out of 14 patients are alive and in remission (21.4%). This highlights the importance of mutational genetic testing in addition to cytogenetic studies to characterize risk stratification and the incorporation of targeted therapy.
Recent studies evaluating different drug combinations and increasing doses have failed to improve outcomes and have increased toxicity in pediatric AML patients [26]. Several new therapeutic agents are currently used in Adult AML patients as targeted therapy [27]. Our results in pediatric AML patients show that molecular analysis, beyond conventional cytogenetic and FISH analysis, must be incorporated for a correct risk stratification and treatment. The incorporation of new agents for targeted therapy associated with conventional chemotherapy schemes is an increasingly urgent need, especially in developing countries like Colombia.
Conclusions
To date, this is the first study in Colombian pediatric AML patients with a complete clinical and genomic characterization. Our study highlights the importance of a rapid and systematic incorporation of genetic analysis in pediatric AML in Colombia, as it directly affects treatment decisions and outcomes.
References
Puumala SE, Ross JA, Aplenc R, Spector LG. Epidemiology of childhood acute myeloid leukemia. Pediatr Blood Cancer. 2013;60(5):728–33.
Conneely SE, Stevens AM. Acute myeloid leukemia in children: emerging paradigms in genetics and new approaches to therapy. Curr Oncol Rep. 2021;23(2):16.
Bravo LE, Garcia LS, Collazos P, Aristizabal P, Ramirez O. Descriptive epidemiology of childhood cancer in Cali: Colombia 1977–2011. Colomb Med (Cali). 2013;44(3):155–64.
CINETS. CNdIeEyTeS. Guía de Práctica Clínica para la detección oportuna, diagnóstico y seguimiento de leucemia linfoide aguda y leucemia mieloide aguda en niños,niñas y adolescentes. Guía No. 9. 2013.
Cheson BD, Bennett JM, Kopecky KJ, Büchner T, Willman CL, Estey EH, et al. Revised recommendations of the international working group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol. 2003;21(24):4642–9.
Creutzig U, Kaspers GJ. Revised recommendations of the International Working Group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol. 2004;22:3432–3.
Common terminology criteria for adverse events (CTCAE) Version 5.0: U.S. Department of health and human services. National institutes of health. national cancer institute. 2017. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_50.
Alexander S, Pole JD, Gibson P, Lee M, Hesser T, Chi SN, et al. Classification of treatment-related mortality in children with cancer: a systematic assessment. Lancet Oncol. 2015;16(16):e604–10.
Arsham MSBM, Lawce HJ. The AGT cytogenetics laboratory manual. 4th ed. New Jersey: Wiley-Blackwell; 2017.
Murphy KM, Levis M, Hafez MJ, Geiger T, Cooper LC, Smith BD, et al. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn. 2003;5(2):96–102.
Verhaak RG, Goudswaard CS, van Putten W, Bijl MA, Sanders MA, Hugens W, et al. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood. 2005;106(12):3747–54.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(3):254–66.
Lin LI, Lin TC, Chou WC, Tang JL, Lin DT, Tien HF. A novel fluorescence-based multiplex PCR assay for rapid simultaneous detection of CEBPA mutations and NPM mutations in patients with acute myeloid leukemias. Leukemia. 2006;20:1899.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, American society of clinical oncology, and college of American pathologists. J Mol Diagn. 2017;19(1):4–23.
Llimpe Y. Cytogenetic risk groups for childhood acute myeloid leukemia based on survival analysis in a cancer referral hospital from Perú. Biomedica. 2021;41(2):302–13.
Deana A, Moran L, Fynn A. Resultados del protocolo GATLA 8-LMAP´07. Nuevos desafíos clínicos en leucemia mieloide aguda pediátrica de novo. Revista Hematología. 2019;23(2):82–91.
Tarlock K, Meshinchi S. Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am. 2015;62(1):75–93.
Bolouri H, Farrar JE, Triche T Jr, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2018;24(1):103–12.
Molina Garay C, Carrillo Sánchez K, Flores Lagunes LL, Jiménez Olivares M, Muñoz Rivas A, Villegas Torres BE, et al. Profiling FLT3 mutations in Mexican acute myeloid leukemia pediatric patients: impact on overall survival. Front Pediatr. 2020;8:586.
Meshinchi S, Woods WG, Stirewalt DL, Sweetser DA, Buckley JD, Tjoa TK, et al. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001;97(1):89–94.
Ishida H, Iguchi A, Aoe M, Nishiuchi R, Matsubara T, Keino D, et al. Panel-based next-generation sequencing facilitates the characterization of childhood acute myeloid leukemia in clinical settings. Biomed Rep. 2020;13(5):46.
Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. 2006;108(12):3654–61.
Kaburagi T, Yamato G, Shiba N, Yoshida K, Hara Y, Tabuchi K, et al. Clinical significance of RAS pathway alterations in pediatric acute myeloid leukemia. Haematologica. 2022;107(3):583–92.
Ishikawa Y, Kawashima N, Atsuta Y, Sugiura I, Sawa M, Dobashi N, et al. Prospective evaluation of prognostic impact of KIT mutations on acute myeloid leukemia with RUNX1-RUNX1T1 and CBFB-MYH11. Blood Adv. 2020;4(1):66–75.
Ayatollahi H, Shajiei A, Sadeghian MH, Sheikhi M, Yazdandoust E, Ghazanfarpour M, et al. Prognostic importance of C-KIT mutations in core binding factor acute myeloid leukemia: a systematic review. Hematol Oncol Stem Cell Ther. 2017;10(1):1–7.
Elgarten CW, Wood AC, Li Y, Alonzo TA, Brodersen LE, Gerbing RB, et al. Outcomes of intensification of induction chemotherapy for children with high-risk acute myeloid leukemia: A report from the children’s oncology group. Pediatr Blood Cancer. 2021;68(12): e29281.
Egan G, Chopra Y, Mourad S, Chiang KY, Hitzler J. Treatment of acute myeloid leukemia in children: a practical perspective. Pediatr Blood Cancer. 2021;68(7): e28979.
Acknowledgements
This work was financed from a grant from Minciencias (Contract 733-2018) to Yunis JJ; Fundación Hospital Pediátrico La Misericordia; Universidad Nacional de Colombia Código Hermes 44702 and Servicios Médicos Yunis Turbay y Cia SAS were all laboratory testing was carried out.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they do not have any conflict of interest to be declared.
Ethical statement
Informed consent was obtained from each patient or guardian. This protocol was approved by the institutional ethics committee of the participant institutions.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Yunis, L.K., Linares-Ballesteros, A., Barros, G. et al. Genomic alterations in a cohort of pediatric acute myeloid leukemia patients at two cancer centers in Colombia. Int J Hematol 117, 269–277 (2023). https://doi.org/10.1007/s12185-022-03475-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-022-03475-w