
Abstract
The decline in the regenerative potential of tissues is one of the most evident characteristics of aging. Stem cell aging determines the aging phenotypes of tissues, and has thus been recognized as one of the hallmarks of mammalian aging. An emerging body of evidence supports an essential role for epigenetic controls in regulating cellular functions. Many epigenetic modifications become stabilized at a particular stage of development. However, epigenetic marks can also readily change over time. This “epigenetic drift” contributes to changes in cellular phenotypes and when it takes place in adult stem cells, may play an important role in stem cell aging. Epigenetic alterations are now recognized as another hallmark of mammalian aging. This process depends on cell intrinsic and extrinsic factors, although the underlying molecular mechanisms remain largely unknown. Here, we review the current progress in the study of epigenetic changes regulating aging hematopoietic stem cells (HSCs). We particularly focus on the epigenome and its regulators in aging HSCs.
Similar content being viewed by others


Introduction
Blood is one of the most highly regenerative tissues, the homeostasis of which is maintained by hematopoietic stem cells (HSCs) throughout the lifetime of the organism. The size of the HSC pool is regulated by a balance between the self-renewal and differentiation of HSCs. HSCs reside in the bone marrow (BM) in adult animals in a specialized microenvironment, the niche. In the BM niche, most HSCs remain in a quiescent state (G0 stage), thereby preserving their capacity for self-renewal. Importantly, however, HSC function declines with age, which causes impaired regeneration of blood system.
The decline in the regenerative potential of tissues is one of the clearest characteristics of aging. Stem cell aging contributes to the aging process in regenerative tissues and together determine their aging phenotypes, thus have been recognized as one of the hallmarks of mammalian aging [1, 2]. HSCs show several characteristic phenotypes with age, such as impaired homing, engraftment potential, and repopulating capacity, and a greater propensity for myeloid differentiation compared with younger HSCs [3–8].
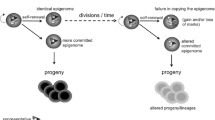
A variety of epigenetic alterations affects all cells and tissues and has been recognized as another hallmark of mammalian aging [1]. Epigenetic regulation of cellular functions is mediated by DNA methylation and histone modifications. The growth and development of higher organisms are regulated by the orchestrated change of epigenetic marks over time, and many epigenetic modifications become stabilized at a particular stage of development. However, epigenetic marks can also readily change over time [9–11]. This “epigenetic drift” is thought to be the result of the stochastic accumulation of epigenetic errors, but also to be induced by acquired somatic mutations in epigenetic regulator genes. The process depends on both intrinsic and extrinsic factors and, when it takes place in adult stem cells, may play an important role in aging, although the underlying molecular mechanisms remain largely unknown [9]. In this review, we focus on the importance of epigenetic regulation in HSC aging.
Epigenetics in HSC aging
Chromatin is composed of nucleosomes, which are molecular complexes of DNA and histone proteins. The term ‘Epigenetics’ is commonly used to describe chromatin-based events, including DNA methylation, histone modification, and chromatin structure. Post-translational modifications of DNA and histones can change chromatin structure and serve as specific sites for reader proteins that recruit additional chromatin-modifying proteins and enzymes. Recently, it has been revealed that non-coding RNAs play a critical role in epigenetic regulation. Epigenetic regulation allows cells to “remember” their gene expression profiles through subsequent cell divisions without any alterations to their DNA sequences.
Epigenetic regulation is required not only for development, but also for tissue homeostasis, which is maintained via the self-renewal and differentiation of somatic stem cells. An accumulating body of evidence suggests that epigenetic regulators play critical roles in the maintenance of self-renewing HSCs [12]. Notably, mutations in epigenetic regulator genes, including genes whose products modify DNA and histones, have been identified in most hematological malignancies [13]. These findings suggest that epigenetic dysregulation can promote the transformation and maintenance of leukemic stem cells (LSCs).
Alterations in epigenome in aging HSCs
Comprehensive DNA methylation profiling of promoter regions in aging mice revealed that nearly a quarter of the queried CpG sites showed age-related methylation changes (both hypermethylation and hypomethylation) in all tissues, with the most prominent changes observed in the most proliferative organs, such as the gastrointestinal tract [14]. Similar proliferation-dependent alterations of the DNA methylation are observed in HSC aging. Changes in DNA methylation during HSC aging occur at regions associated with lineage potential and selectively target genes exclusively expressed downstream of HSCs or expressed at higher levels in progenitor and/or effector cells relative to HSCs. In addition, polycomb group (PcG) protein targets in embryonic stem cells are enriched among hypermethylated genes [15]. These findings suggest that stochastic DNA methylation drift (gradual increases or decreases at specific loci) creates epigenetic mosaicism in aging stem cells that may restrict their plasticity and worsen phenotypes such as stem cell exhaustion and focal proliferative defects that can lead to cancer [9].
Age-associated global hypomethylation has been observed in mouse tissues [16] and human cells, such as peripheral leukocytes [17], although the changes were quite small; the loss of the mean 5-methylcytosine (5-mC) content in aged leukocytes being 2 % of that in the young adult leukocytes [17]. Whole-genome bisulfite sequencing (WGBS) of 4-month-old (young) and 14-month-old (aged) HSCs, however, showed that DNA methylation increases in HSCs with age from 83.5 to 84.6 % [18]. In the same study, alterations of the DNA methylation were similarly observed in the context of HSC aging (both hypermethylation and hypomethylation) and it again appeared that polycomb-targeted genes are hot spots for both hyper- and hypo-differentially methylated CpGs related to aging. Interestingly, 70 % of the HSC fingerprint genes, which are associated with HSC maintenance [19], showed hypomethylation that correlated with their increased expression with age. In addition, hyper-differentially methylated regions are highly enriched for the binding sites of Pu.1, a key regulator of HSC differentiation, mutation or deletion of which is closely associated with leukemogenesis [20]. All of these changes may reinforce HSC self-renewal, diminish differentiation, and increase the risk of transformation, paralleling phenotypic HSC aging behavior. Unexpectedly, this study also revealed that ribosomal biogenesis is a prominent target of aging, with increased transcription of ribosomal protein and RNA genes and hypomethylation of rRNA genes. Although ribosomal biogenesis has been linked to aging in model organisms such as yeast and worms, its role in mammalian aging warrants careful examination [21].
Genome-wide comparisons of histone modifications between young and aged mouse HSCs have also provided potential mechanisms that contribute to HSC aging [18]. Aged HSCs exhibited broader H3K4me3 peaks, particularly in HSC identity and self-renewal genes, showing a positive correlation with gene expression changes. Changes in H3K27me3 levels have also been described in aging. Expression of the PcG protein Ezh2 declines in aging islet β cells, resulting in reduced levels of H3K27me3 associated with de-repression of the Ink4a/Arf locus [22]. H3K27me3 levels decrease strikingly with age throughout the tissues of C. elegans, and RNA interference of an H3K27me3 demethylase UTX-1 has been shown to extend lifespan in these worms [23]. These findings suggest that organismal aging is associated with a reduction in H3K27me3 levels in differentiated tissues. In contrast, levels of H3K27me3 are low across the genome in quiescent adult skeletal muscle stem cells in young mice, but this repressive chromatin mark accumulates and spreads with age [24]. In aged HSCs, H3K27me3 peak counts does not significantly change in ChIP-sequence analysis between young and aged mouse HSCs, but the average signal intensity at the transcription start site (TSS) shows a trend to increase [18]. These alterations in the aging HSC epigenome are summarized in Table 1.
Role of PcG proteins in HSC aging
Two major pathways implicated in the induction of senescence involve tumor suppressors including p16Ink4a and p19Arf (p14ARF in human) that are encoded in the Ink4a/Arf (Cdkn2a) locus (Fig. 1). p19Arf stabilizes p53 protein by inhibiting MDM2, which mediates ubiquitin-dependent degradation of p53, resulting in the activation of p53 target genes involved in cell cycle arrest and apoptosis such as p21. p16Ink4a blocks the assembly of catalytically active cyclin D–CDK4/6 complexes, thereby keeping retinoblastoma protein (Rb) hypophosphorylated. Rb then represses E2F-dependent transcription by sequestering E2F, ultimately leading to cell cycle arrest and senescence. p21 associates with and inhibits the cyclin E/A–CDK2 complex, linking the p53 and Rb pathways [25]. HSCs are exposed to various stresses, such as telomere attrition, DNA damage, oxidative stress, and activation of oncogenes. Any of these stresses may induce expression of p16Ink4a and p19Arf in HSCs leading to the cell cycle arrest, senescence, or apoptosis. Sustained activation of these tumor suppressors eventually causes depletion of HSCs and regulates HSC aging [2, 26, 27]. Indeed, their expression and/or function increases with age [28]. p16Ink4a, which accumulates with age, accounts, at least in part, for age-dependent changes in islet β cells, forebrain neural progenitors, HSCs, and T lymphocytes [29–32], whereas the impact of p19Arf has not been tested in aging tissues. Strict regulation of these tumor suppressors thus appears to hold a key to maintaining HSC function over the lifetime of an animal and preventing premature HSC aging.

Pathways regulating aging of HSCs. a Organization of the Ink4b–Arf–Ink4a locus. Boxes denote exons. A part of exon 2 is translated in alternative reading frames. b Effects of Ink4a and Arf tumor suppressor pathways. p16Ink4a blocks the assembly of catalytically active cyclin D-CDK4/6 complexes, thereby keeping Rb hypophosphorylated. Rb then represses E2F-dependent transcription by sequestering E2F, ultimately leading to G1-phase cell cycle arrest and senescence. p19Arf (p14ARF in human) stabilizes the p53 protein by inhibiting ubiquitin ligase activity of MDM2, which promotes polyubiquitination and ubiquitin-dependent degradation of p53, resulting in the activation of p53 target genes involved in G1-phase cell cycle arrest and apoptosis such as p21. p21 associates with and inhibits the cyclin E/A-CDK2 complex, linking the p53 and Rb pathways
Stem cells have developed epigenetic regulatory mechanisms to prevent premature aging. PcG genes are representative of such regulators. PcG proteins form various polycomb repressive complexes (PRC) (Fig. 2). The PRC1 and PRC2 complexes possess H2AK119 ubiquitin ligase activity and H3K27 methyltransferase activity, respectively. Two PcG complexes function in both cooperative and independent manners to establish and maintain gene silencing. Polycomb complexes are general regulators of stem cells, and play an essential role in the maintenance of HSC function [12, 33, 34]; Bmi1, a component of PRC1, plays a central role in this process. Bmi1-deficient mice exhibit a severe defect in HSCs accompanied by marked de-repression of p16 Ink4a and p19 Arf [35, 36]. Importantly, deletion of both Ink4a and Arf substantially restores the impaired self-renewal capacity of Bmi1 −/− HSCs [37]. De-repression of p16 Ink4a and p19 Arf has also been observed in HSCs deficient for PRC2 genes, Ezh1 and Eed, and deletion of both Ink4a and Arf is again effective to rescue impaired HSC function at least partially [38, 39]. All these findings suggest that PcG complexes regulate HSCs by acting as a critical failsafe against premature aging induced by p16Ink4a and p19Arf tumor suppressors. Conversely, forced expression of Bmi1, Ezh2, Cbx7, and Kdm2b/Fbxl10 genes promotes HSC self-renewal and efficiently preserves HSC potential during serial transplantations, suggestive of their role in HSC aging [40–43]. Despite this growing body of evidence supporting the role of PcG complexes in HSC aging, we still do not know how changes in PcG complex activity contribute to HSC aging other than by its impact on the Ink4a/Arf locus.

Maintenance of transcriptional repression of target genes by Polycomb repressive complexes. The PcG proteins assemble into large multiprotein complexes, the best-characterized being PRC1 and PRC2. RING1, a component of PRC1 catalyzes H2AK119 ubiquitylation (H2AK119ub1) and promotes chromatin compaction and transcriptional repression. The EZH component of PRC2 contains a SET domain and catalyzes the methylation of lysine 27 of histone H3. H3K27me3 serves as a binding site for CBX and thereby mediates recruitment of the PRC1 complex to PcG target gene loci. Recently, Eed-deficient mouse ES cells showed only a minor decrease in H2Aub1 levels despite a drastic reduction in H3K27me3 [77], suggesting a role for PRC2-independent RING1-containing complexes which lack CBX component (non-canonical PRC1: PRC1.1-1.6), which rely on RYBP/YAF2, L3MBTL2 or KDM2B for their recruitment to CpG-rich promoters [33]
Roles of sirtuins in HSC aging
Members of the sirtuin family of NAD+-dependent protein deacetylases, particularly Sir2, have been studied extensively as potential anti-aging factors. However, its role in worms and flies has recently become controversial [44]. In mammals, there are seven Sir2 homologs (sirtuins). They have discrete subcellular localizations, with a subset of sirtuins residing in predominantly nuclear (SIRT1, SIRT6, and SIRT7), cytosolic (SIRT2), or mitochondrial (SIRT3, SIRT4, and SIRT5) compartments. The Sir2 family of histone deacetylases (HDACs) targets H4K16 and also various proteins, including those involved in the regulation of glucose and fatty acid metabolisms [45, 46] (Fig. 3). Notably, levels of acetylation at H4K16 decrease with age in both human and mouse tissues, including HSCs [47, 48]. Pharmacological inhibition of elevated activity of the small RhoGTPase Cdc42 in aged HSCs functionally rejuvenates aged HSCs, and restores the level and spatial distribution of histone H4K16 acetylation to a status similar to that seen in young HSCs. These findings indicate that epigenetic regulation by Sir2 family HDACs governed by the Cdc42 activity plays a role in HSC aging, although the detailed mechanisms underlying this activity remain undetermined [48].

Role of sirtuins in epigenetics. Sirtuins are a class of HDACs that require nicotinamide adenine dinucleotide (NAD+). Their activity may therefore depend on nutritional and metabolic factors. They deacetylate histones, such as H4K16Ac, and other proteins in a reaction that consumes NAD+. Sirt3 is a unique family member that localizes to mitochondria and regulates the acetylation status of mitochondrial proteins, such as IDH2. Hyper-caloric diets cause a low NAD+/NADH ratio and, consequently, low sirtuin activity. Conversely, calorie restriction causes a high NAD+/NADH ratio and therefore increases the sirtuin activity
Mutant mice deficient in Sirt6 exhibit accelerated aging [49], whereas male transgenic mice overexpressing Sirt6 have a longer lifespan than control animals [50]. In hematopoietic stem and progenitor cells (HSPCs), loss of Sirt1, which is the closest homolog to invertebrate Sir2, causes an increase in Hoxa9 expression and expansion of HSPCs under conditions of hematopoietic stress, which results in increased accumulation of DNA damage and exhaustion of HSPCs in mice [51]. In contrast, the mitochondrial sirtuin Sirt3 regulates the global acetylation landscape of mitochondrial proteins. For example, Sirt3 directly deacetylates and activates mitochondrial isocitrate dehydrogenase 2 (IDH2), leading to increased NADPH levels and an increased ratio of reduced-to-oxidized glutathione in mitochondria [52]. Importantly, Sirt3, which is enriched in HSCs and suppressed with aging, is dispensable for HSCs at a young age under homeostatic conditions, but is essential under hematopoietic stress and in old age. In addition, overexpression of Sirt3 improves the regenerative capacity of aged HSCs [53]. These findings indicate that sirtuins contribute to the regulation of HSC aging by regulating both histone modifications and mitochondrial function.
Epigenetics in acquisition of clonal hematopoiesis with age
Approximately, 11,000 HSCs reside in the human bone marrow, of which only 1,300 are active, indicating that most of the HSCs are quiescent [54]. The clonal composition of the HSC pool changes significantly during a lifetime and consists of several types of HSCs with different self-renewal capacity and differentiation potential. Aging HSCs typically show myeloid-biased differentiation and increased incidence of myeloid malignancies [55, 56]. Correspondingly, the fraction of cells with inactivation of each X chromosome shows age-associated skewing (AAS) in human female blood cells, particularly in myeloid-lineage cells [57, 58], suggesting a change in stem cell usage with stochastic loss of some of the original stem cells with age. It has been reported that HSC quiescence and concomitant attenuation of DNA repair and response pathways underlies DNA damage accumulation in HSCs during aging, providing a potential mechanism by which premalignant mutations accumulate in HSCs [59]. Notably, inactivating somatic TET2 mutations were identified in granulocyte DNA but not in lymphocyte or buccal DNA in 5.6 % of normal elderly females with acquired clonal hematopoiesis defined by X-inactivation skewing [60]. Tet2 deletion or haploinsufficiency results in enhanced HSC self-renewal and myeloid-biased hematopoiesis in mice [61, 62]. These findings suggest that TET2 mutation in HSCs trigger clonal expansion of HSCs that exhibit myeloid-biased differentiation, contributing to the emergence of aging hematopoietic phenotypes. TET proteins are mammalian DNA hydroxylases that catalyze the conversion of 5-mC to 5-hydroxymethylcytosine (5-hmC). TET proteins then perform iterative oxidation of 5-hmC to produce 5-formylcytosine (5-fC) followed by 5-carboxylcytosine (5-caC). Through these steps, TET proteins promote DNA demethylation [63]. Quantitative mass spectrometry revealed reduced 5-hmC levels in mouse aged HSCs [18] as well as human peripheral blood cells with age [60], indicating reduced 5-hmC is an epigenetic hallmark of aging hematopoietic cells, with acquired TET2 mutations being one mechanism responsible for the reduction.
Transformation of aged HSCs associated with epigenetic dysregulation
There are strong interactions between aging, genetic and epigenetic alterations, and disease. Aging, chronic inflammation, and environmental insults are major factors that alter the epigenome, and indeed cancer cells have various epigenetic alterations [64]. A majority of CpG islands methylated in colon cancer are also methylated in normal colon during the process of aging [65, 66]. A similar trend is observed in myelodysplastic syndrome (MDS) [67]. These findings suggest that epigenetic changes in aging closely track with cancer risk, and are thus likely associated with an early step in transformation.
Recent deep-sequencing data reinforced long-standing hypotheses about the stem cell origins of cancer [68] and the clonal evolution of cancers through Darwinian selection [69]. As described in the previous section, DNA damage accumulates in stem cells during aging, increasing the risk of acquisition of premalignant mutations in stem cells [59]. Importantly, genetic alterations often involve epigenetic regulator genes that encode histone, nucleosome remodeling proteins, and DNA and histone modifying proteins, suggesting that the downstream epigenetic defects are functionally linked to the neoplastic phenotype [13, 70, 71]. Of these, recurrent mutations in genes that regulate DNA methylation including DNMT3A, TET2, IDH1, and IDH2 have been identified in pre-leukemic stem cells (pre-LSCs). Acquisition of these mutations in HSCs can cause their clonal expansion resulting in a pre-LSC population. Pre-LSCs retain the ability to differentiate into all kinds of mature progeny, but can become fully transformed with the acquisition of additional driver mutations [72, 73]. Thus, these mutations are thought to have an initiating role in the pathogenesis of myeloid malignancies. These recent advances have revealed a link between genetic alterations and epigenetic changes in all types of tumors.
Future perspectives
To date, only some epigenetic marks have been shown to alter with age in HSCs. Particularly, only a very few comprehensive analyses have been reported for histone modifications in aged HSCs. The detailed epigenetic mechanisms of stem cell aging thus remain largely unknown. However, HSCs lacking histone modification enzymes, such as PcG proteins and SIRT deacetylases, show aging-related phenotypes. Conversely, overexpression of some histone modification enzymes prevents HSC aging. These findings strongly implicate epigenetic regulation in HSC aging.
Epigenetic drift is thought to depend both on intrinsic and external environmental factors. Although HSC aging has been thought to be caused primarily by factors intrinsic to the HSC compartment, environmental factors, including chemical pollutants, dietary components, and other external stresses can have long-lasting effects on the epigenome [74]. Furthermore, recent data highlight an important role for HSC-extrinsic factors in promoting HSC aging [8], including changes in niche composition or function and systemic factors that are secreted at distal sites and circulated throughout the body. Growth differentiation factor 11 (GDF11) is such a circulating protein, which can slow the age-dependent deterioration of the neurogenic niche as well as that in skeletal muscle [75, 76]. However, the link between the epigenetic regulators and external factors is largely missing. The function of epigenetic regulators is regulated by chemical modifications such as phosphorylation and cellular-level substrates required for the epigenetic enzymes to exert their catalytic activity. The means by which external signals, including those from niches and systemic circulation, affect the function of epigenetic regulators in HSCs are important undiscovered mechanisms. Much work remains to be done to decipher the complete picture of epigenetic machineries that regulate HSC aging.
References
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217.
Signer RA, Morrison SJ. Mechanisms that regulate stem cell aging and life span. Cell Stem Cell. 2013;12:152–65.
Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2:1011–6.
Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000;192:1273–80.
Kamminga LM, van Os R, Ausema A, Noach EJ, Weersing E, Dontje B, et al. Impaired hematopoietic stem cell functioning after serial transplantation and during normal aging. Stem Cells. 2005;23:82–92.
Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. 2011;208:2691–703.
Geiger H, Denkinger M, Schirmbeck R. Hematopoietic stem cell aging. Curr Opin Immunol. 2014;29C:86–92. doi:10.1016/j.coi.2014.05.002.
Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013;13:376–89.
Issa JP. Aging and epigenetic drift: a vicious cycle. J Clin Invest. 2014;124:24–9.
Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23:413–8.
Han S, Brunet A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012;22:42–9.
Sashida G, Iwama A. Epigenetic regulation of hematopoiesis. Int J Hematol. 2012;96:405–12.
Chung YR, Schatoff E, Abdel-Wahab O. Epigenetic alterations in hematopoietic malignancies. Int J Hematol. 2012;96:413–27.
Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–40.
Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013;12:413–25.
Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem. 1987;262:9948–51.
Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, Miura K, Miyazaki T, et al. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Ann Hum Genet. 2004;68:196–204.
Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–88.
Chambers SM, Boles NC, Lin KY, Tierney MP, Bowman TV, Bradfute SB, Chen AJ, et al. Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell. 2007;1:578–91.
Koschmieder S, Rosenbauer F, Steidl U, Owens BM, Tenen DG. Role of transcription factors C/EBPα and PU.1 in normal hematopoiesis and leukemia. Int J Hematol. 2005;81:368–77.
Fontana L, Partridge L, Longo VD. Extending healthy life span-from yeast to humans. Science. 2010;328:321–6.
Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009;23:975–85.
Maures TJ, Greer EL, Hauswirth AG, Brunet A. The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell. 2011;10:980–90.
Liu L, Cheung TH, Charville GW, Hurgo BM, Leavitt T, Shih J, et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4:189–204.
Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–77.
Newgard CB, Sharpless NE. Coming of age: molecular drivers of aging and therapeutic opportunities. J Clin Invest. 2013;123:946–50.
Pollina EA, Brunet A. Epigenetic regulation of aging stem cells. Oncogene. 2011;30:3105–26.
Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–307.
Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4A induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–7.
Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–52.
Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–6.
Liu Y, Johnson SM, Fedoriw Y, Rogers AB, Yuan H, Krishnamurthy J, et al. Expression of p16Ink4a prevents cancer and promotes aging in lymphocytes. Blood. 2011;117:3257–67.
Laugesen A, Helin K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell. 2014;14:735–51.
Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299–313.
Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–5.
Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21:843–51.
Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, Nakauchi H. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J Exp Med. 2006;203:2247–53.
Hidalgo I, Herrera-Merchan A, Ligos JM, Carramolino L, Nunez J, Martinez F, et al. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell. 2012;11:649–62.
Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014;14:68–80.
Nakamura S, Oshima M, Yuan J, Saraya A, Miyagi S, Konuma T, et al. Bmi1 confers resistance to oxidative stress on hematopoietic stem cells. PLoS One. 2012;7:e36209.
Kamminga LM, Bystrykh LV, de Boer A, Houwer S, Douma J, Weersing E, et al. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006;107:2170–9.
Klauke K, Radulović V, Broekhuis M, Weersing E, Zwart E, Olthof S, et al. Polycomb Cbx family members mediate the balance between haematopoietic stem cell self-renewal and differentiation. Nat Cell Biol. 2013;15:353–62.
Konuma T, Nakamura S, Miyagi S, Negishi M, Chiba T, Oguro H, et al. Forced expression of the histone demethylase Fbxl10 maintains self-renewing hematopoietic stem cells. Exp Hematol. 2011;39:697–709.
Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvári M, Piper MD, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011;477:482–5.
Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–38.
Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family’s role in aging and age-associated pathologies. J Clin Invest. 2013;123:973–9.
Krishnan V, Chow MZ, Wang Z, Zhang L, Liu B, Liu X, et al. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci USA. 2011;108:12325–30.
Florian MC, Dörr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10:520–30.
Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–29.
Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–21.
Singh SK, Williams CA, Klarmann K, Burkett SS, Keller JR, Oberdoerffer P. Sirt1 ablation promotes stress-induced loss of epigenetic and genomic hematopoietic stem and progenitor cell maintenance. J Exp Med. 2013;210:987–1001.
Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–12.
Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, et al. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013;3:319–27.
Catlin SN, Busque L, Gale RE, Guttorp P, Abkowitz JL. The replication rate of human hematopoietic stem cells in vivo. Blood. 2011;117:4460–6.
Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci USA. 2010;107:5465–70.
Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci USA. 2011;108:20012–7.
Busque L, Mio R, Mattioli J, Brais E, Blais N, Lalonde Y, et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65.
Gale RE, Fielding AK, Harrison CN, Linch DC. Acquired skewing of X-chromosome inactivation patterns in myeloid cells of the elderly suggests stochastic clonal loss with age. Br J Haematol. 1997;98:512–9.
Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell. 2014;. doi:10.1016/j.stem.2014.04.016.
Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44:1179–81.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24.
Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y, et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med. 2013;210:2627–39.
Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68.
Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–27.
Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–94.
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–6.
Maegawa S, Gough SM, Watanabe-Okochi N, Lu Y, Zhang N, Castoro RJ, et al. Age-related epigenetic drift in the pathogenesis of MDS and AML. Genome Res. 2014;24:580–91.
Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200.
Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8.
Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55.
Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–33.
Chan SM, Majeti R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int J Hematol. 2013;98:648–57.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109.
Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344:630–4.
Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–52.
Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 2010;24:265–76.
Acknowledgments
This work was supported in part by a grants-in-aid for Scientific Research (#24249054) and Scientific Research on Innovative Areas ‘Stem cell aging and disease’ (#26115002) from MEXT, Japan.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Oshima, M., Iwama, A. Epigenetics of hematopoietic stem cell aging and disease. Int J Hematol 100, 326–334 (2014). https://doi.org/10.1007/s12185-014-1647-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-014-1647-2