
Abstract
Breaking water-in-oil emulsions during the refining of crude oils is an important step before any upgrading process is started. Asphaltene molecules are incriminated as playing an important role in this phenomenon. Unraveling the mechanisms behind the affinity between them and water is a key step to understand how to break these emulsions more easily and require lower amounts of demulsifiers. Choosing which demulsifier molecule(s) to use is also primordial, but to do so rationally, one needs to know which are the molecular interactions in place between asphaltenes, porphyrins and water so that demulsifiers are chosen to destabilize a specific physical–chemical interaction. In this paper, we study the interactions arising between asphaltenes and porphyrins and six different molecules potentially displaying a demulsification action in the presence of water/oil interfaces. We demonstrate that the ionic demulsifier molecules present an interesting potential to either interact strongly with water, replacing asphaltenes in this interaction, or to interact with the active sites of asphaltenes, deactivating them and avoiding any asphaltenic interfacial activity. Finally, we also found that although asphaltenes do not migrate spontaneously toward the water/oil interfaces, porphyrins do so rather easily. This indicates that porphyrins do have an important activity at the water/oil interface.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Before crude oil can be refined, dissolved and suspended salts must be removed. This step is known as the desalting phase and consists of adding water to the crude, yielding often water-in-oil (W/O) emulsions (Varadaraj and Brons 2012). Breaking them is a fundamental step in order to avoid corrosion, to reduce pumping cost due to an increased viscosity, among others (Masliyah et al. 2011; Adams 2014; Gawel et al. 2005; Marchal et al. 2010; Kilpatrick 2012).
Asphaltenes are traditionally seen as hydrophobic molecules constituting of a conjugated core grafted with lateral chains (Dutta Majumdar et al. 2013). Controversially, they are also indicated by numerous studies to play a role in the stabilization of W/O emulsions (McLean and Kilpatrick 1997; Yan et al. 1999; Yarranton et al. 2000; Gafonova and Yarranton 2001; Kilpatrick 2012; Shi et al. 2017; Spiecker et al. 2003a, b), which could only be possible if one accepts the presence of polar groups containing heteroatoms or metals, besides their apolar conjugated core (Varadaraj and Brons 2012).
Moreover, these polar groups would also play an important role in the construction process of asphaltenes aggregates via the formation of hydrogen bonds among them (Carbognani 2000; Dudášová et al. 2008; Adams 2014, Teklebrhan et al. 2016). We have recently showed the impact of these H-bonds on the formation of nanoaggregates and how this fact can be considered a game-changer for the model describing this challenging physical–chemical process (Santos Silva et al. 2018). These results led us to propose that the traditional asphaltene aggregation model (Yen-Mullins) (Mullins et al. 2012) may be very well adapted for some types of molecules, and it should be a particular case of a more general, supramolecular, model, as proposed by Gray et al. (2011). Beyond our own work, other authors have also shown the validity of the Gray model to describe the complex structures formed between asphaltenes and water at the water/oil interface (Spiecker and Kilpatrick 2004; Yarranton et al. 2007a, b; Verruto et al. 2009; Sedghi et al. 2013; Mikami et al. 2013; Gao et al. 2014; Liu et al. 2015).
Since asphaltenes are defined as a class of solubility (SARA fractionation Jewell et al. 1972) rather than a class of molecules with specific properties, only a fraction of the molecules may have emulsifying properties (concomitant presence of hydrophobic and hydrophilic groups), while other fractions are inert (Jarvis et al. 2015; Czarnecki and Moran 2005; Czarnecki 2008; Yang et al. 2014; Wu 2003). As a result, asphaltenes in general may be wrongly identified as being the only ones responsible for the undesirable stability of crude oil emulsions formed during the desalting process (Kilpatrick 2012; Qiao et al. 2017 and the references therein). Such a limitation is, undoubtedly, detrimental to the thorough understanding of these mechanisms.
Qiao et al. (2017) proposed a novel fractionation method consisting in separating the asphaltenes on the basis of their adsorption properties at the water/oil interfaces. The fraction showing significant interfacial activity was estimated to be only 2% in the weight of the total amount of isolated asphaltenes. The analysis of this fraction shows that its molecular mass is higher and the amounts of carbon, hydrogen, nitrogen and sulfur are similar to those found in asphaltenes not demonstrating such activity. On the other hand, the amount of oxygen is 3 times higher and can be attributed, thanks to the FTIR analysis (band around 1700 cm−1), according to the study, to a more important number of sulfoxides (Yang et al. 2015), carboxylic acids, carbonyls and their derivatives (Qiao et al. 2017). Once these sub-fractions are separated out, the remaining asphaltenes show almost no emulsion formation and stabilization, thus corroborating the results already found by Yang et al. (2014). Similar results were also found by Ligiero et al. (2017) after a careful analysis by mass spectroscopy coupled with size-exclusion chromatography of the interfacial material.
The easiest way to model an amphiphilic behavior in asphaltenes without inserting too many suppositions is by studying the impact of carboxylic acids on these molecules. These can be present in crude oil in two forms: naphthenic acids, non-aromatic structures which are mostly found in heavy and immature oils, their origin being indicated as being due to the biodegradation of microorganisms (Meredith et al. 2000; Brandal et al. 2006); or as end groups of asphaltenes’ side chains, which gives them an amphiphilic character (Varadaraj and Brons 2007). By revealing their presence in the fraction responsible for the stabilization of emulsions, we can also immediately see the link with the corrosion of pipelines due to emulsions: The interaction between these acids from the emulsion and the chromium(III) oxide from the surface of stainless steel can also contribute to this unwanted process (Qiao et al. 2017).
Previously, we showed that the presence of these acids also supports the hypothesis of a supramolecular aggregation of asphaltenes (Santos Silva et al. 2017, 2018). In fact, when not ionized, these groups can form H-bonds networks, stabilizing cluster of nanoaggregates or water droplets, as already indicated by other authors (Kilpatrick 2012). Experimental evidence shows that a few molecules containing this group suffice to considerably impact the behavior of a complex whole of molecules (Liu et al. 2015).
Concerning the water/oil interfaces, it has been hypothesized that acid/base interactions within crude oil may be behind the stabilization of emulsions (Fortuny et al. 2007; Kilpatrick 2012; Subramanian et al. 2017). Molecules capable of forming a network of interactions around water molecules, including hydrogen bonds, are also indicated as being possible targets to reduce such stabilization process (Kilpatrick 2012; Qiao et al. 2017).
Despite all these advances, some questions concerning the role played by asphaltenes at these interfaces remain: Do they migrate spontaneously? What is the role played by porphyrins? Do demulsifier molecules act preferably on which one of them? How this activity is perturbed by different thermodynamic conditions? etc. Given the cost that emulsion breaking imposes to the refining production chain, understanding the emulsion destabilization process is of utmost importance, particularly if one is capable of pinpointing the molecular origin of this process. Setting the conditions for the demulsification process is nowadays considered by many as a “work of art” (Dimova et al. 2017) since so little is known on how the molecular structures responsible for emulsion formation interact with other chemicals and thermodynamic conditions.
Motivated by this, we have performed molecular dynamics simulations on asphaltene systems in order to model the interface region and its dependance on several parameters, such as: the presence of demulsifiers, porphyrins, different thermodynamic conditions, and electric fields. Our aim remains to pinpoint the molecular origin of a given behavior in models just complex enough to be representative of the chemical diversity found experimentally.
2 Methodology
First off, it is worth saying that simulating explicit emulsions is a complex task since they are basically thermodynamically unstable but kinetically stable (Lissant 1988; Sjöblom 2001). Even if the diffusion times of the asphaltene molecules toward the interfaces can be very short, in the scale of molecular modeling, this represents very long simulation times (Liu et al. 2015). It is for this reason that this work is not intended to be a complete study of the formation and destabilization of emulsions, but rather to highlight important physical–chemical interactions under (d-)emulsifying conditions.
The simulation boxes studied in this work are constituted of a mixture of asphaltenes (10 different molecules, 5 molecules of each type—vide infra) and porphyrins (5 vanadyl and 5 nickel, 3 of each containing a polar lateral chain and 2 of each having only a small apolar lateral chain—vide infra). This system was solvated with a mixture of toluene (6417 molecules), n-heptane (900 molecules—154,8 g/L) and water (320 molecules—10 g/L). The so-formed solutions have an asphaltene concentration on the order of ~ 5 wt%. Full details on how these conditions were set can be found in our previous work (Santos Silva et al. 2018).
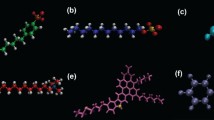
The studied asphaltene molecules are based on the so-called CA22 (Fig. 1a) molecule identified by Schuler et al. (2015), to which we have grafted two lateral chains on the opposite sides of the molecule. Then, the length of this chain was set to be n-C6H13 or n-C16H33 (Jian et al. 2013), and the number of fused rings on the conjugated core was set to 7 or 11, as seen in Fig. 1a, b. The so-called PA3 molecule type described by these authors was also studied with two different lateral chains. These molecules are defined as presented in Table 1 alongside the labeling system used. The reasons that led to choosing these molecules, in particular, were thoroughly discussed in our last papers (Santos Silva et al. 2016, 2017). Finally, it is important to recall that oxygen concentration for these asphaltenes systems is of the order of 1.5%, whereas reported values in literature are around 1% (Rane et al. 2012).

Molecular structure of the studied molecules with increasing conjugated core size and lateral chain length. a Molecules of the type AAX (derived from CA22), having 25 π-conjugated carbon atoms; b ADX molecules, 35 π-conjugated carbon atoms. Finally, c the PA3-type molecule, having 32 π-conjugated carbon atoms
The studied porphyrin molecules are presented in Fig. 2. Two different cases were envisaged: porphyrins having no polar lateral chains and porphyrins having a chain. The force field parameters of the metal center were obtained by a density functional theory–Hessian fitting performed by an in-house code called mammoth. One can found the so-obtained topologies as a separate SI file.

Molecular structure of the porphyrin molecules herein studied: a nickel porphyrin having no polar lateral chain; b vanadyl porphyrin having no polar lateral chain; c nickel porphyrin having a polar lateral chain; and d vanadyl porphyrin having a polar lateral chain
The simulation box hereafter called “reference” was created by introducing all these molecules randomly in a 12 × 12 × 12 nm3 simulation box. The energy of such system was minimized by steepest descendant and conjugated gradient methods. Then, it was equilibrated during 3 ns using a NPT ensemble (V-rescale (Bussi et al. 2007) thermostat and Berendsen et al. (1984) barostat) set at 298 K and 1 bar. The configuration issued from this phase was submitted to a NPT ensemble [Nosé-Hoover (Hoover 1985; Nosé 1984) thermostat and Parrinello-Rahman (Parrinello and Rahman 1981) barostat] at the same T, P conditions during 60 ns. Atomic velocities were initiated using a Maxwell–Boltzmann distribution. During all simulations, van der Waals and Coulomb interactions were cut off at 1.2 nm. Periodic boundary conditions were applied in the three directions. The Verlet algorithm (Verlet 1967) was used for integration of the Newton equations using a timestep of 2 fs. Electrostatic interactions were calculated using particle mesh Ewald method (Essmann et al. 1995) in a FFT grid with 0.16-nm spacing. Bond vibrations were constrained using the LINCS algorithm (Hess et al. 1997). All neighbor lists were constructed taking into account particles within a 1.2-nm sphere and updated at each 5 steps. All these simulations were performed in Gromacs (Van Der Spoel et al. 2005) package, version 5.1. The topologies for all the molecules in this study were obtained using the PRODRG server (Schüttelkopf and Van Aalten 2004) to obtain parameters for the GROMOS96-53a6 force field (van Gunsteren et al. 2002). Water molecules were described using the SPC/E model (Berendsen et al. 1987).
To this “reference” configuration, several other simulations were performed. First of all, a given quantity of a molecule having a potential role as a demulsifier was also added to this system and the minimization–stabilization–production phases were started over again. As several molecules with demulsifying properties exist, we have picked some representative systems from the literature with some (d)emulsifying potential: ethyl cellulose (Feng et al. 2008; Feng et al. 2011), sulfonic acids (Chang and Fogler 1994) and salts of tetra-alkylammonium (Standnes and Austad 2000; Austad and Standnes 2003; Salehi et al. 2008). Particularly, these salts have been shown to be able to interact with the strongly absorbed anionic carboxylates on the liquid–solid interfaces in the reservoirs. The ionic pairs thus formed aid in the desorption of the organic material of the interface (Pons-Jiménez et al. 2015). This same behavior can also be found for analogous but anionic surfactants, even if the mechanism is not entirely elucidated and the yields are lower. Motivated by this, six molecules of demulsifiers were chosen (see Fig. 3 for molecular structures), which are: Tween20™ (TW2); Span20™ (SP2); S-limonene (SLM); ethyl cellulose (EC); hexadecyltrimethylammonium chloride (cetyltrimethylammonium chloride—CTAC); 1-hexadecanesulfonate sodium salt (HSAN). These molecules were chosen to study their anionic, cationic, neutral, long or short chain behavior, etc. More particularly, S-limonene was chosen since it is an abundant waste material from paper industry and display excellent solvation properties.

Molecular structure of molecules displaying a potential demulsifier character: a Tween20; b Span20; c S-limonene; d ethyl cellulose; e hexadecyltrimethylammonium chloride; and f 1-hexadecanesulfonate sodium salt
The same process was repeated, but now we also added a water bilayer around the simulation box containing other 18,318 additional water molecules. In order to determine the quantity of such demulsifier molecules, we have taken into account the mass of the full system containing the water bilayer. To respect a meaningful ratio of around 1200 ppm, one would need 1 molecule of Tween20, 4 of Span20, 10 of SLM, 3 of EC, 4 of CTAC and 4 of HSAN. EC was considered as being 3 monomers each molecule. The counter ions were set free for the case of the ionic molecules.
The resulting asphaltenic systems were submitted to the conditions found in the desalting/demulsifying processes in the oil’s refining chain: Besides ambient thermodynamic conditions, we have also considered T = 423 K and P = 15 bar. These systems had three electric fields configurations (none, 1500 V/cm parallel or 1500 V/cm perpendicular to the interface), yielding a total of 30 different systems. The motivation behind the study of electric fields is due to the fact that numerous advantages can be attributed to them when breaking W/O emulsions: reduction in the charge in chemicals, apparatus simplicity, high yields, etc. (Kwon et al. 2010). The action mechanism is attributed to the perturbation of electrostatic interaction within the emulsion, yielding an accelerated coalescence of water droplets (Eow et al. 2001). Even though no free salt is present in our systems, considering any possible electric fields allows us to pinpoint their responsibility on the emulsion-breaking process.
3 Results and discussion
3.1 Demulsifier–water interactions
Before screening the effect of demulsifiers on asphaltene–water interactions, we studied their behavior in a toluene/n-heptane/water solvent system. From the analysis of the mean squared displacement (MSD) and the resultant diffusion coefficients (D), as well as radial distribution functions (RDFs) and interaction energies, one can verify that as expected, SLM interacts the least with water, followed by EC and TW2. In contrast, ionic molecules (CTAC and HSAN) as well as SP2 have reduced diffusion coefficients, indicating a higher degree of interaction with water. Although SP2 and TW2 have very similar molecular structures, the SP2 interacts considerably more with water than the TW2, possibly due to the fact that the latter can form intramolecular hydrogen bonds, whereas this is not possible for SP2 (see Fig. S.9 and S.10). This was also confirmed by the analysis of H-bonds interactions throughout the simulations, indicating that TW2 indeed almost does not form such interaction with the water molecules but rather does with itself.
3.2 Asphaltene systems in the presence of a water/oil interface under ambient thermodynamic conditions
3.2.1 No demulsifier
The analysis of the RDFs, MSD, interaction energies and H-bond formation for asphaltene–asphaltene interactions shows that their level of aggregation increases with the presence of a water/oil interface. Under these conditions, they form 2 times more H-bonds than the situation where water is only present in the form of a droplet (Fig. S.8). This fact suggests that asphaltenes prefer to interact among them rather than with the interface.
After 60 ns of simulation, asphaltenes do not migrate spontaneously toward the interface. On the other hand, the water droplet is wrapped by the asphaltenes via hydrogen bonds, intermediated by the carboxylic acid groups grafted at the end of the side chains of asphaltenes (see Fig. 4). Porphyrins also seems to play a role. In the presence of the interface, porphyrins migrate to the aqueous layer. In our simulations, these porphyrins were PVO (vanadyl porphyrin without side chain, green) and PNC (porphyrin nickel with polar side chain, magenta). These observations indicate that vanadyl or nickel porphyrins with polar side chains probably play an important role in any interfacial activity that may present the asphaltene phase.

Snapshots after 60 ns of simulation. Toluene and n-heptane molecules, although present in all simulations, are not shown. The porphyrins of vanadyl and nickel without polar side chains are shown in green and gray, respectively. If they have these chains, they are represented in ocher and magenta, respectively. Demulsifier molecules are presented in bright red. From top to bottom: TW2, SLM, HSAN

Snapshots after 60 ns of simulation. Toluene and n-heptane molecules, although present in all simulations, are not shown. The porphyrins of vanadyl and nickel without polar side chains are shown in green and gray, respectively. If they have these chains, they are represented in ocher and magenta, respectively. Demulsifier molecules are presented in bright red. From top to bottom: CTAC, EC, SP2
3.2.2 With demulsifier
We present in this section the complete asphaltenic systems consisting of a mixture of asphaltenes, porphyrins, toluene, n-heptane, water and demulsifiers. Two scenarios were studied: systems with or without an interface.
From the analysis of D (Fig. S.2), one can see that SLM has no interaction with other molecules, asphaltenes included, regardless of the presence of an interface. On the other hand, the other molecules tested are more or less likely to present such interactions. CTAC (cationic) and TW2 (neutral) have the lowest diffusion coefficient, while HSAN (anionic) and EC (neutral) have intermediate values and SP2 (neutral) has a higher value. In the presence of an interface, the conclusion for SLM is maintained, whereas for the other molecules, the observed trends are not the same as for the case without interface. The origin of this behavior is studied in more detail in the following paragraphs. Figures 4 and 5 present end-of-simulation snapshots for the situations with and without aqueous interfaces.
Based on the analysis of RDFs (Fig. S.3), asphaltene–asphaltene interactions at the short distance region (nanoaggregation) are unchanged in the presence of an interface (Ruiz-Morales and Mullins 2015). On the other hand, long-range interactions (inter-nanoaggregates) are impacted by the presence of interfaces, characterized by an increase in their intensity due to spatial constraint. Concerning demulsifiers, their presence induces an increase in the interaction between nanoaggregates, as it can be noted from the analysis of the CN/N (%) ratio. For recall, this index is obtained from the ratio between the integrals of the first peak and the whole RDF function for a given system. In this way, it characterizes the proportion of π-stacks over all the other possible interactions existing between asphaltenes in the simulation box. Such effect, observed for either interface-related systems, is maximized when the demulsifier is the SP2 molecule type. The origins behind this effect are scrutinized in the following section.
Asphaltene–porphyrins interactions are around 15% less intense when there is an water/oil interface (see Fig. S.4, probably a sign of a preference of either one or the other to interact with this region of the simulation box). When demulsifiers are also present, such interactions are also lowered, probably due to the fact that demulsifiers interacting with porphyrins and/or asphaltenes reduce the interaction among these species and water, as it was recently indicated by Jiang et al. (2019).
On the other hand, remarkably for the situation without an interface, asphaltene–water interactions are around 66% less intense when demulsifiers are present, except when it is SLM, as it can be noted from Fig. S.5. This indicates that demulsifiers interact with either asphaltenes or water in order to hinder their interaction.
More interestingly, porphyrin–water interactions are considerably more sensible to the presence of an interface. With no demulsifier, the presence of an interface induces a ~ fourfold increase (Fig. S.6) in the intensity of this interaction. This is a clear indication that with an interface, porphyrins have an increased tendency to migrate to these regions of the simulation box, which can also be clearly noted from the snapshots presented in Fig.s 4 and 5. In the same line, for the case of no interface, the presence of demulsifiers lowers the porphyrin–water interaction by about 50%, by either bonding directly to the water droplet or to the porphyrin molecule. With an interface, the presence of demulsifiers passes unnoticed, very probably because they are in a much lower concentration compared to the increased quantity of water.
Among the asphaltene–demulsifier interactions, the most intense ones are those formed with CTAC, HSAN or SP2 molecules, which can be noted from both the analyses of interaction energies and H-bonds (Figs. S.7 and S.11). In the presence of an interface, these interactions are considerably lower, indicating that demulsifiers prefer to interact with water, more abundant in this situation.
Porphyrins’ preferable demulsifiers are CTAC, HSAN and SP2 for both situations where there is a water/oil interface or not. For the situation without interface, it is interesting to note that CTAC has a greater interaction than HSAN. Since this first molecule is cationic, it probably interacts more easily with the partial charge on the oxygen atom of the vanadyl group of such porphyrins, whereas HSAN must interact directly with the metal, whose positive charge (either nickel or vanadium) is much more difficult to access (exposition to the solution) than the oxygen of the vanadyl group, or with the few polar lateral groups.
In the next section, these observations will be confronted to the presence of distinct thermodynamic conditions and electric fields.
3.3 Asphaltene systems in the presence of a water/oil interface and demulsifiers under desalting conditions
In this section, we focus on systems having an explicit water/oil interface and submit them to different thermodynamic conditions and electric fields corresponding to those used in desalting. Among the six demulsifier molecules studied in the previous section, only five are hereafter considered: SLM not presenting any interactions with neither asphaltenes nor water, it has been excluded. The final configurations of the previous simulations were used as starting points of novel 60-ns molecular dynamics runs. This was also the case for the ambient conditions.
Taking into account the presence of an electric field is tricky since its effects are heavily linked to the presence and concentration of dissolved salts (electrolytes) in the medium. However, in the framework of this work, we decided to consider no electrolyte in order to verify that electric fields alone have no impact on the aggregation structures of asphaltenes under such conditions.
Figure S.12 presents the diffusion coefficients of asphaltenes under these conditions. It is clear that asphaltene is more mobile at desalting than ambient thermodynamic conditions, regardless of the electric fields and the demulsifiers. Porphyrins behave similarly, even if the increase in this coefficient is around 6 times (Fig. S.13), indicating that porphyrins are much more mobile at such conditions than asphaltenes. The same was also observed for demulsifiers, as it can be seen in Fig. S.14. Compared to both asphaltenes and porphyrins, demulsifiers are the molecules with the highest mobilities in the simulation box. In order to determine particular interactions, each pair of interacting molecule types is studied in more detail in the next paragraphs.
The increase in temperature and pressure, regardless of the electric field, induces an increase in the CN/N (%) ratio (Fig. S.15) for asphaltene–asphaltene interactions. This could be due to the fact that at such conditions, asphaltene’s mobility is increased. Consequently, being more mobile, nanoaggregates have an increased probability of forming connections among them via H-bonds, namely. On the other hand, the interaction energies are almost unchanged under desalting conditions compared to ambient ones (Fig. S.16). This is a clear sign that nanoaggregation is not sensible to this pressure/temperature range.
Interestingly, the opposite is observed for asphaltene–porphyrins interactions: There is indeed a reduction in their intensity when one passes from 298 K/1 bar to 423 K/15 bar conditions, regardless of the presence of electric fields, as it can be deducted from Fig. 6. This means that while asphaltene nanoaggregates interact more at this temperature, the interactions between asphaltenes and porphyrins are more fragile and can be than released from nanoaggregates.

Asphaltene–porphyrin interaction energies in the presence of demulsifier molecules, various thermodynamic conditions and electric fields. PRL and PPD mean parallel and perpendicular to the water/oil interface, respectively
Asphaltene–demulsifiers interactions are the most intense when the former are HSAN and SP2. Under desalting conditions, CTAC molecules are straightforwardly carried to the interface, reducing the intensity of its interaction with asphaltenes. H-bonds interactions are observed for HSAN molecules via the sulfate oxygen atoms. Generally speaking, asphaltene–demulsifier H-bonds are favored at a higher temperature, as it can be deducted from Fig. 7.

Total number of H-bonds between asphaltenes and demulsifiers in the presence of demulsifier molecules, various thermodynamic conditions and electric fields

Final configurations after 60 ns of simulation. Toluene and n-heptane molecules, although present in all simulations, are not shown. The vanadyl and nickel-bearing porphyrins without polar side chains appear in green and gray, respectively. If they have these chains, they are represented, respectively, in ocher and magenta. The demulsifier is presented in bright red

Final configurations after 60 ns of simulation. Toluene and n-heptane molecules, although present in all simulations, are not shown. The vanadyl and nickel-bearing porphyrins without polar side chains appear in green and gray, respectively. If they have these chains, they are represented, respectively, in ocher and magenta. The demulsifier is presented in bright red

Final configurations after 60 ns of simulation. Toluene and n-heptane molecules, although present in all simulations, are not shown. The vanadyl and nickel-bearing porphyrins without polar side chains appear in green and gray, respectively. If they have these chains, they are represented, respectively, in ocher and magenta. The demulsifier is presented in bright red. This zoom shows the Span20 demulsifier in action, in interaction with the aqueous layer, with porphyrins or with asphaltenes

Final configurations after 60 ns of simulation. Toluene and n-heptane molecules, although present in all simulations, are not shown. The vanadyl and nickel-bearing porphyrins without polar side chains appear in green and gray, respectively. If they have these chains, they are represented, respectively, in ocher and magenta. The demulsifier is presented in bright red. This zoom shows the action of porphyrins as interfacial agents: By binding to nanoaggregates of asphaltenes, they can lead them to interfaces. In addition, these molecules can also efficiently structure nanoaggregates, bringing together several of them
SP2 molecules, in particular, display strong H-bonds interactions with asphaltenes which are also magnified under desalting conditions, probably due to a coupled mechanism of increased diffusion of the demulsifier and the possibility of forming stable H-bonds with asphaltenes. As this molecule has only a little affinity with water compared to its ionic counterparts, it might be more freely available to interact with asphaltenes at the oily phase. Despite the structural similarities shared with TW2, TW2 only forms a fraction of these interactions with asphaltenes. This is due to the fact that TW2 is capable of forming intramolecular H-bonds and becomes in this way inactive toward interactions with asphaltenes.
On the other hand, porphyrin–demulsifier interactions are more strong with HSAN molecules rather than SP2 ones, as it can be noted from Figs. S.20–22. In this figure, one can clearly note that these are H-bond interactions and no other demulsifier molecules present consistent interactions with porphyrins, even for the case of CTAC, despite its cationic character. One would expect strong electrostatic interactions between the ammonium anion and the vanadyl group, but this seems not to be the case. As aforementioned, this is probably due to the fact that CTAC molecules are straightforwardly attracted by the water/oil interfaces, being unavailable in the organic phase.
Asphaltene–water interactions are disadvantaged by the high-temperature conditions: There is a reduction of around 25% of its intensity when one passes from 298 K/1 bar to 423 K/15 bar (Figs. S.17 and S.18). This effect is also observed from the analysis of H-bonds, which are more frequent at low temperatures. This indicates that the desalting conditions contribute to the reduction in the interaction between asphaltenes and the interfaces. The same is also observed for porphyrin–water interactions, for which the reduction is now around 50% (Fig. S.19), and the mechanism is similar to the one taking place for asphaltenes: The H-bonds interactions between porphyrins and water are decreased by a factor of two when desalting conditions are used. For both interactions (asphaltenes–water and porphyrins–water), no dependence on the electric field intensity and orientation could be observed.
3.4 Review of key details
Having presented the effect of both demulsifiers and desalting thermodynamic conditions on specific molecular interactions that could be responsible for the stabilization of water/oil interfaces, it is interesting to make a recall of the key observations issued from our simulations. The following figures present the end-of-simulation snapshots of such complex asphaltene systems under such conditions. They allow one to highlight the following points (Figs. 8, 9, 10, 11):
- a.
The complex asphaltenic systems studied under different conditions have in common the formation of more or less extended, Gray-like aggregates. Porphyrins migrate spontaneously toward the water/oil interface, and this effect can be somewhat reversed under desalting conditions.
- b.
Among the different demulsifiers studied, CTAC, HSAN and SP2 are the most active. Particularly, HSAN can interact with water, porphyrins and asphaltenes molecules. It can be verified that the sulfate group interacts with the asphaltenes via their side chains. Also, this demulsifier can destabilize H-bonds between asphaltenes and water.
- c.
Similarly, SP2 molecules can form H-bonds with water, porphyrins and asphaltenes molecules. The attraction of this molecule to the polar groups of asphaltenes can be clearly seen, preventing them from making such interactions with other molecules.
- d.
Porphyrins may carry asphaltenes to the interface and also stabilize their self interactions in these regions. Also, vanadium porphyrins with side chains can be easily identified structuring several nanoaggregates together, stabilizing the hydrogen interactions between them and thus being a likely clustering and flocculation agent.
4 Conclusions
In this paper, we have studied the interactions between different demulsifiers and water; the role of water/oil interfaces on the aggregation of a mixture of asphaltene molecules; the asphaltene–interfaces and porphyrins–interfaces interactions; and the demulsifier–asphaltene, and demulsifiers–porphyrins interactions. These were also studied under desalting thermodynamic conditions and submitted to electric field perpendicular or parallel to the water/oil interface. This approach, intended to replicate the operational conditions found at the refinery, is a first attempt to identify key parameters to be monitored and/or pursued in future studies.
The presence of the interface does not have a major impact on the nanoaggregation of asphaltenes, but inter-nanoaggregate interactions (macro-aggregation/clusterization) are impacted by these interfaces since nanoaggregates are forced to interact more with each other. At high temperatures, water is much more soluble in toluene and spreads throughout the simulation box. Despite this, we could not observe the trapping of these molecules within nanoaggregates. Also, asphaltene nanoaggregates are more or less extended, displaying supramolecular interactions characteristics of a Gray-like model.
Our findings do not indicate any effect due to the electric fields, regardless of its polarity, thermodynamic condition or demulsifier presence. This is probably due to its too low intensity to be considered in a conventional molecular dynamics study Dimova et al. (2017), as well as the absence of electrodes and electrolytes in the aqueous phase (dissolved salts).
Under low and high temperatures, porphyrins migrate easily to the water/oil interfaces, whereas this is not the case for asphaltenes. The asphaltenes, if found in this region, were carried there by the porphyrins. For these porphyrins, the interactions with water are mediated by hydrogen bonds made with their side chains and/or the vanadyl group. Moreover, porphyrins can also be easily inserted into nanoaggregates of asphaltenes via the formation of hydrogen bonds. These molecules can thus “crosslink” this phase, i.e., bind several nanoclusters together.
Among the demulsifiers studied in this step, those with the most notable asphaltene and porphyrin interaction potential are HSAN, CTAC and SP2. These molecules can interact via hydrogen bonds or electrostatic interactions with the polar groups of asphaltenes, inactivating them. Moreover, CTAC molecule also displays interesting interactions: They have the possibility to interact with ionized carboxylic groups (not presented in this paper), inactivating it, besides competing strongly with asphaltenes on the interaction with water molecules, making it more difficult for them to present an interfacial activity. In addition, the role played by HSAN via the inactivation of the polar group of asphaltenes was also observed at the water/oil interface, whereas as SP2 is preferred by the oily phase, it plays its role when the asphaltenes are not yet on these problematic areas. Future studies should utilize mixtures of HSAN and Span20 as well as CTAC and Span20 to correlate their demulsifier properties with the acidity character of the crude oil at the origin of the emulsion.
References
Adams JJ. Asphaltene adsorption, a literature review. Energy Fuels. 2014;28(5):2831–56. https://doi.org/10.1021/ef500282p.
Austad T, Standnes DC. Spontaneous imbibition of water into oil-wet carbonates. J Pet Sci Eng. 2003;39(3–4):363–76. https://doi.org/10.1016/S0920-4105(03)00075-5.
Berendsen HJ, van Postma JPM, van Gunsteren WF, DiNola ARHJ, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–90. https://doi.org/10.1063/1.448118.
Berendsen HJC, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J Phys Chem. 1987;91(24):6269–71. https://doi.org/10.1021/j100308a038.
Brandal Ø, Hanneseth A-MD, Hemmingsen P, Sjöblom J, Kim S, Rodgers RP, et al. Isolation and characterization of naphthenic acids from a metal naphthenate deposit: molecular properties at oil-water and air-water interfaces. J Dispersion Sci Technol. 2006;27(3):295–305. https://doi.org/10.1080/01932690500357909.
Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1):014101. https://doi.org/10.1063/1.2408420.
Carbognani L. Effects of iron compounds on the retention of oil polar hydrocarbons over solid sorbents. Pet Sci Technol. 2000;18(3–4):335–60. https://doi.org/10.1080/10916460008949850.
Chang C-L, Fogler HS. Stabilization of asphaltenes in aliphatic solvents using alkylbenzene-derived amphiphiles. 2. Study of the asphaltene-amphiphile interactions and structures using Fourier transform infrared spectroscopy and small-angle X-ray scattering techniques. Langmuir. 1994;10(6):1758–66. https://doi.org/10.1021/la00018a022.
Czarnecki J. Stabilization of water in crude oil emulsions. Part 2. Energy Fuels. 2008;23(3):1253–7. https://doi.org/10.1021/ef800607u.
Czarnecki J, Moran K. On the stabilization mechanism of water-in-oil emulsions in petroleum systems. Energy Fuels. 2005;19(5):2074–9. https://doi.org/10.1021/ef0501400.
Dimova D, Pisov S, Panchev N, Nedyalkova M, Madurga S, Proykova A. Insight into electric field-induced rupture mechanism of water-in-toluene emulsion films from a model system. J Chem Phys. 2017;146(19):194703. https://doi.org/10.1063/1.4983163.
Dudášová D, Simon S, Hemmingsen PV, Sjöblom J. Study of asphaltenes adsorption onto different minerals and clays: part 1. Experimental adsorption with UV depletion detection. Colloids Surf A Physicochem Eng Asp. 2008;317(1–3):1–9. https://doi.org/10.1016/j.colsurfa.2007.09.023.
Dutta Majumdar R, Gerken M, Mikula R, Hazendonk P. Validation of the Yen–Mullins model of athabasca oil-sands asphaltenes using solution-state 1H Nmr relaxation and 2D Hsqc spectroscopy. Energy Fuels. 2013;27(11):6528–37. https://doi.org/10.1021/ef401412w.
Eow JS, Ghadiri M, Sharif AO, Williams TJ. Electrostatic enhancement of coalescence of water droplets in oil: a review of the current understanding. Chem Eng J. 2001;84(3):173–92. https://doi.org/10.1016/S1385-8947(00)00386-7.
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–93. https://doi.org/10.1063/1.470117.
Feng X, Wang S, Hou J, Wang L, Cepuch C, Masliyah J, et al. Effect of hydroxyl content and molecular weight of biodegradable ethylcellulose on demulsification of water-in-diluted bitumen emulsions. Ind Eng Chem Res. 2011;50(10):6347–54. https://doi.org/10.1021/ie102071q.
Feng X, Zhenghe X, Masliyah J. Biodegradable polymer for demulsification of water-in-bitumen emulsions. Energy Fuels. 2008;23(1):451–6. https://doi.org/10.1021/ef800825n.
Fortuny M, Oliveira CB, Melo RL, Nele M, Coutinho RC, Santos AF. Effect of salinity, temperature, water content, and pH on the microwave demulsification of crude oil emulsions. Energy Fuels. 2007;21(3):1358–64. https://doi.org/10.1021/ef0603885.
Gafonova OV, Yarranton HW. The stabilization of water-in-hydrocarbon emulsions by asphaltenes and resins. J Colloid Interface Sci. 2001;241(2):469–78. https://doi.org/10.1006/jcis.2001.7731.
Gao F, Zhen X, Liu G, Yuan S. Molecular dynamics simulation: the behavior of asphaltene in crude oil and at the oil/water interface. Energy Fuels. 2014;28(12):7368–76. https://doi.org/10.1021/ef5020428.
Gawel I, Bociarska D, Biskupski P. Effect of asphaltenes on hydroprocessing of heavy oils and residua. Appl Catal A. 2005;295(1):89–94. https://doi.org/10.1016/j.apcata.2005.08.001.
Gray MR, Tykwinski RR, Stryker JM, Tan X. Supramolecular assembly model for aggregation of petroleum asphaltenes. Energy Fuels. 2011;25(7):3125–34. https://doi.org/10.1021/ef200654p.
Hess B, Bekker H, Berendsen HJ, Fraaije JG. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–72. https://doi.org/10.1002/(SICI)1096-987X(199709)18:12%3c1463:AID-JCC4%3e3.0.CO;2-H.
Hoover WG. Canonical dynamics: equilibrium phase-space distributions. Phys Rev A. 1985;31(3):1695. https://doi.org/10.1103/PhysRevA.31.1695.
Jarvis JM, Robbins WK, Corilo YE, Rodgers RP. Novel method to isolate interfacial material. Energy Fuels. 2015;29(11):7058–64. https://doi.org/10.1021/acs.energyfuels.5b01787.
Jewell DM, Weber JH, Bunger JW, Plancher H, Latham DR. Ion-exchange, coordination, and adsorption chromatographic separation of heavy-end petroleum distillates. Anal Chem. 1972;44(8):1391–5. https://doi.org/10.1021/ac60316a003.
Jian C, Tang T, Bhattacharjee S. Probing the effect of side-chain length on the aggregation of a model asphaltene using molecular dynamics simulations. Energy Fuels. 2013;27(4):2057–67. https://doi.org/10.1021/ef400097h.
Jiang B, Zhang R, Yang N, Zhang L, Sun Y, Jian C, et al. Molecular mechanisms of suppressing asphaltene aggregation and flocculation by dodecylbenzenesulfonic acid probed by molecular dynamics simulations. Energy Fuels. 2019;33(6):5067–80. https://doi.org/10.1021/acs.energyfuels.9b00821.
Kilpatrick PK. Water-in-crude oil emulsion stabilization: review and unanswered questions. Energy Fuels. 2012;26(7):4017–26. https://doi.org/10.1021/ef3003262.
Kwon W-T, Park K, Han SD, Yoon SM, Kim JY, Bae W, et al. Investigation of water separation from water-in-oil emulsion using electric field. J Ind Eng Chem. 2010;16(5):684–7. https://doi.org/10.1016/j.jiec.2010.07.018.
Ligiero LM, et al. Characterization of crude oil interfacial material isolated by the wet silica method. Part 1: gel permeation chromatography inductively coupled plasma high-resolution mass spectrometry analysis. Energy Fuels. 2017;31(2):1065–71. https://doi.org/10.1021/acs.energyfuels.6b02899.
Lissant KJ. Emulsification and demulsification: an historical overview. Colloids Surf. 1988;29(1):1–5. https://doi.org/10.1016/0166-6622(88)80168-9.
Liu J, Zhao Y, Ren S. Molecular dynamics simulation of self-aggregation of asphaltenes at an oil/water interface: formation and destruction of the asphaltene protective film. Energy Fuels. 2015;29(2):1233–42. https://doi.org/10.1021/ef5019737.
Mammoth: Model for Atomic Molecular Mechanics on the Hub. 2018. https://mammoth_uppa.gitlab.io.
Marchal C, Abdessalem E, Tayakout-Fayolle M, Uzio D. Asphaltene diffusion and adsorption in modified Nimo alumina catalysts followed by ultraviolet (UV) spectroscopy. Energy Fuels. 2010;24(8):4290–300. https://doi.org/10.1021/ef1000979.
Masliyah JH, Xu Z, Czarnecki JA. Handbook on theory and practice of bitumen recovery from Athabasca oil sands. Theoretical basis, vol. 1. Denver: Kingsley Knowledge Publishing; 2011.
McLean JD, Kilpatrick PK. Effects of asphaltene aggregation in model heptane-toluene mixtures on stability of water-in-oil emulsions. J Colloid Interface Sci. 1997;196(1):23–34. https://doi.org/10.1006/jcis.1997.5177.
Meredith W, Kelland S-J, Jones DM. Influence of biodegradation on crude oil acidity and carboxylic acid composition. Org Geochem. 2000;31(11):1059–73. https://doi.org/10.1016/S0146-6380(00)00136-4.
Mikami Y, Liang Y, Matsuoka T, Boek ES. Molecular dynamics simulations of asphaltenes at the oil-water interface: from nanoaggregation to thin-film formation. Energy Fuels. 2013;27(4):1838–45. https://doi.org/10.1021/ef301610q.
Mullins OC, Sabbah H, Eyssautier J, Pomerantz AE, Barré L, Andrews AB, Ruiz-Morales Y, et al. Advances in asphaltene science and the Yen–Mullins model. Energy Fuels. 2012;26(7):3986–4003. https://doi.org/10.1021/ef300185p.
Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol Phys. 1984;52(2):255–68. https://doi.org/10.1080/00268978400101201.
Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52(12):7182–90. https://doi.org/10.1063/1.328693.
Pons-Jiménez M, Cisneros-Dévora R, Gómez-Balderas R, Cartas-Rosado R, Oviedo-Roa R, Beltrán HI, et al. Supramolecular pairing among heteroaromatic compounds and the cationic surfactant C12tac. Fuel. 2015;149:174–83.
Qiao P, Harbottle D, Tchoukov P, Masliyah J, Sjöblom J, Liu Q, et al. Fractionation of asphaltenes in understanding their role in petroleum emulsion stability and fouling. Energy Fuels. 2017;31(4):3330–7. https://doi.org/10.1021/acs.energyfuels.6b02401.
Rane JP, Harbottle D, Pauchard V, Couzis A, Banerjee S. Adsorption kinetics of asphaltenes at the oil–water interface and nanoaggregation in the bulk. Langmuir. 2012;28:9986–95. https://doi.org/10.1021/la301423c.
Ruiz-Morales Y, Mullins OC. Coarse-grained molecular simulations to investigate asphaltenes at the oil–water interface. Energy Fuels. 2015;29(3):1597–609. https://doi.org/10.1021/ef502766v.
Salehi M, Johnson SJ, Liang J-T. Mechanistic study of wettability alteration using surfactants with applications in naturally fractured reservoirs. Langmuir. 2008;24(24):14099–107. https://doi.org/10.1021/la802464u.
Santos Silva H, Alfarra A, Vallverdu G, Bégué D, Bouyssiere B, Baraille I. Sensitivity of asphaltene aggregation toward the molecular architecture under desalting thermodynamic conditions. Energy Fuels. 2017. https://doi.org/10.1021/acs.energyfuels.7b02728.
Santos Silva H, Alfarra A, Vallverdu G, Bégué D, Bouyssiere B, Baraille I. Impact of H-bonds and porphyrins on asphaltene aggregation as revealed by molecular dynamics simulations. Energy Fuels. 2018;32(11):11153–64. https://doi.org/10.1021/acs.energyfuels.8b01901.
Silva S, Hugo AC, Sodero BB, Carrier H, Korb J-P, Alfarra A, et al. Molecular dynamics study of nanoaggregation in asphaltene mixtures: effects of the N, O, and S heteroatoms. Energy Fuels. 2016;30(7):5656–64. https://doi.org/10.1021/acs.energyfuels.6b01170.
Schuler B, Meyer G, Peña D, Mullins OC, Gross L. Unraveling the molecular structures of asphaltenes by atomic force microscopy. J Am Chem Soc. 2015;137(31):9870–6. https://doi.org/10.1021/jacs.5b04056.
Schüttelkopf AW, Van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(8):1355–63. https://doi.org/10.1107/S0907444904011679.
Sedghi M, Goual L, Welch W, Kubelka J. Effect of asphaltene structure on association and aggregation using molecular dynamics. J Phys Chem B. 2013;117(18):5765–76. https://doi.org/10.1021/jp401584u.
Shi C, Zhang L, Xie L, Xi L, Liu Q, He J, et al. Surface interaction of water-in-oil emulsion droplets with interfacially active asphaltenes. Langmuir. 2017;33(5):1265–74. https://doi.org/10.1021/acs.langmuir.6b04265.
Sjöblom J. Encyclopedic handbook of emulsion technology. Boca Raton: CRC Press; 2001. https://doi.org/10.1201/9780367801281.
Spiecker PM, Gawrys KL, Kilpatrick PK. Aggregation and solubility behavior of asphaltenes and their subfractions. J Colloid Interface Sci. 2003a;267(1):178–93. https://doi.org/10.1016/S0021-9797(03)00641-6.
Spiecker PM, Gawrys KL, Trail CB, Kilpatrick PK. Effects of petroleum resins on asphaltene aggregation and water-in-oil emulsion formation. Colloids Surf A. 2003b;220(1–3):9–27. https://doi.org/10.1016/S0927-7757(03)00079-7.
Spiecker PM, Kilpatrick PK. Interfacial rheology of petroleum asphaltenes at the oil–water interface. Langmuir. 2004;20(10):4022–32. https://doi.org/10.1021/la0356351.
Standnes DC, Austad T. Wettability alteration in chalk: 2. Mechanism for wettability alteration from oil-wet to water-wet using surfactants. J Pet Sci Eng. 2000;28(3):123–43.
Subramanian D, May N, Firoozabadi A. Functional molecules and the stability of water-in-crude oil emulsions. Energy Fuels. 2017;31(9):8967–77. https://doi.org/10.1021/acs.energyfuels.7b01039.
Teklebrhan RB, Jian C, Choi P, Zhenghe X. Competitive adsorption of naphthenic acids and polyaromatic molecules at a toluene–water interface. J Phys Chem B. 2016;120(50):12901–10. https://doi.org/10.1021/acs.jpcb.6b07938.
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–18. https://doi.org/10.1002/jcc.20291.
van Gunsteren WF, Daura X, Mark AE. GROMOS force field. In encyclopedia of computational chemistry. Chichester: Wiley; 2002. https://doi.org/10.1002/0470845015.cga011.
Varadaraj R, Brons C. Molecular origins of crude oil interfacial activity part 3: characterization of the complex fluid rag layer formed at crude oil–water interfaces. Energy Fuels. 2007;21(3):1617–21. https://doi.org/10.1021/ef0606299.
Varadaraj R, Brons C. Molecular origins of crude oil interfacial activity part 4: oil-water interface elasticity and crude oil asphaltene films. Energy Fuels. 2012;26(12):7164–9. https://doi.org/10.1021/ef300830f.
Verlet L. Computer experiments on classical fluids. I. Thermodynamical properties of Lennard–Jones molecules. Phys Rev. 1967;159(1):98.
Verruto VJ, Le RK, Kilpatrick PK. Adsorption and molecular rearrangement of amphoteric species at oil-water interfaces. J Phys Chem B. 2009;113(42):13788–99. https://doi.org/10.1021/jp902923j.
Wu X. Investigating the stability mechanism of water-in-diluted bitumen emulsions through isolation and characterization of the stabilizing materials at the interface. Energy Fuels. 2003;17(1):179–90. https://doi.org/10.1021/ef020098y.
Yan Z, Elliott JA, Masliyah JH. Roles of various bitumen components in the stability of water-in-diluted-bitumen emulsions. J Colloid Interface Sci. 1999;220(2):329–37. https://doi.org/10.1006/jcis.1999.6533.
Yang F, Tchoukov P, Dettman H, Teklebrhan RB, Liu L, Dabros T, et al. asphaltene subfractions responsible for stabilizing water-in-crude oil emulsions. Part 2: molecular representations and molecular dynamics simulations. Energy Fuels. 2015;29(8):4783–94. https://doi.org/10.1021/acs.energyfuels.5b00657.
Yang F, Tchoukov P, Pensini E, Dabros T, Czarnecki J, Masliyah J, et al. Asphaltene subfractions responsible for stabilizing water-in-crude oil emulsions. Part 1: Interfacial behaviors. Energy Fuels. 2014;28(11):6897–904. https://doi.org/10.1021/ef501826g.
Yarranton HW, Hussein H, Masliyah JH. Water-in-hydrocarbon emulsions stabilized by asphaltenes at low concentrations. J Colloid Interface Sci. 2000;228(1):52–63. https://doi.org/10.1006/jcis.2000.6938.
Yarranton HW, Sztukowski DM, Urrutia P. Effect of interfacial rheology on model emulsion coalescence: I. Interfacial rheology. J Colloid Interface Sci. 2007a;310(1):246–52. https://doi.org/10.1016/j.jcis.2007.01.071.
Yarranton HW, Urrutia P, Sztukowski DM. Effect of interfacial rheology on model emulsion coalescence: II. Emulsion coalescence. J Colloid Interface Sci. 2007b;310(1):253–9. https://doi.org/10.1016/j.jcis.2007.01.098.
Acknowledgements
The authors thank the DN (Direction du Numérique) from Université de Pau et des Pays de l’Adour, MCIA (Mésocentre de Calcul Intensif Aquitain) and GENCI-CINES (Grant 2017-c2016087698) for providing the computation power needed for this project. Isifor-Carnot Institute and Total Refining & Chemicals are also acknowledged for their financial support to this research project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Xiu-Qiu Peng
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Santos Silva, H., Alfarra, A., Vallverdu, G. et al. Role of the porphyrins and demulsifiers in the aggregation process of asphaltenes at water/oil interfaces under desalting conditions: a molecular dynamics study. Pet. Sci. 17, 797–810 (2020). https://doi.org/10.1007/s12182-020-00426-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12182-020-00426-0