
Abstract
Purpose of Review
Hypertension is a common disorder with substantial impact on public health due to highly elevated cardiovascular risk. The mechanisms still remain unclear and treatments are not sufficient to reduce risk in majority of patients. Inflammatory mechanisms may provide an important mechanism linking hypertension and cardiovascular risk. We aim to review newly identified immune and inflammatory mechanisms of hypertension with focus on their potential therapeutic impact.
Recent Findings
In addition to the established role of the vasculature, kidneys and central nervous system in pathogenesis of hypertension, low-grade inflammation contributes to this disorder as indicated by experimental models and GWAS studies pointing to SH2B3 immune gene as top key driver of hypertension. Immune responses in hypertension are greatly driven by neoantigens generated by oxidative stress and modulated by chemokines such as RANTES, IP-10 and microRNAs including miR-21 and miR-155 with other molecules under investigation. Cells of both innate and adoptive immune system infiltrate vasculature and kidneys, affecting their function by releasing pro-inflammatory mediators and reactive oxygen species.
Summary
Immune and inflammatory mechanisms of hypertension provide a link between high blood pressure and increased cardiovascular risk, and reduction of blood pressure without attention to these underlying mechanisms is not sufficient to reduce risk.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypertension (HT) is a common disorder with substantial impact on public health because of its potential sequelae of stroke, heart failure and kidney disease; subsequently, it is a major source of morbidity and mortality [1]. It affects 30% of adults in Europe and USA with additional 30% at high risk of the disease [2], and importantly, its prevalence increases with age [3].
Despite the progress in its diagnosis and treatment, the aetiology of HT remains unclear and a matter of substantial debate. It is widely acknowledged that function of the vascular system, kidneys and sympathetic nervous system is critical for control and maintenance of blood pressure (BP) [1]. Vascular resistance, stiffness and remodelling as well endothelial dysfunction are hallmarks of HT [4,5,6,7]. Kidney transplantation from hypertensive donors rises BP in recipients in animal and human studies [8, 9]. Sympathetic nervous system hyperactivity contributes to initiation, maintenance and progression of HT [10]. Furthermore, deletion of extracellular superoxide dismutase or administration of angiotensin II (Ang II) into circumventricular organs (CVO) raises blood pressure, while lesions of these regions prevent experimental HT [11, 12].
Inflammation—Hallmark of Hypertension
In addition to established roles of the vasculature, kidneys and central nervous system, there is a mounting evidence, to suggest that chronic low-grade inflammation contributes to cardiovascular disease (CVD) [13••, 14], including HT [15]. For instance, C-reactive protein (CRP) is a prototypic marker of inflammation which is elevated in many CVD such as acute myocardial infraction, coronary artery disease and HT [13••, 16]. Vongpatanasin et al. have shown that modest elevations in CRP are sufficient not only to increase BP but also exaggerate response to Ang II [17]. Furthermore, human and animal studies emphasize impact of CRP on endothelial vasodilator function [18, 19] via direct action on endothelial nitric oxide synthase (eNOS) and/or activation of vascular NADPH oxidases leading to reactive oxygen species (ROS) formation [19,20,21]. In addition to CRP, the inflammatory process involves a complex interplay between cells and pro-inflammatory cytokines. This process is strictly controlled by the immune system. Various cells of the innate and adaptive immune systems contribute to initiation and maintenance of inflammation. Furthermore, resolution of inflammation occurs by different mechanism in many CVD, including HT.
Link Between Immune Cells and Hypertension
The concept that the immune system contributes to hypertension is not new. Fifty years ago, the pioneering study of White and Grollman described the role of immunosuppressive therapy on BP levels in rats with kidney infarction [22]. Further studies have shown that the transfer of immune cells isolated from lymph nodes or spleen of hypertensive animals increases BP in normotensive recipients [23, 24]. Moreover, thymectomy of deoxycorticosterone acate (DOCA)-salt-treated mice [25] or spontaneously hypertensive rats [26] attenuates experimental HT. Despite the above-mentioned studies showing the potential role of the immune system in HT, lack of advanced animal models and methodology did not allow for a more thorough understanding this phenomenon.
T Cells
In 2007, the landmark study by Guzik et al. demonstrated that HT induced by Ang II or DOCA salt is blunted in RAG1−/− mice lacking functional lymphocytes, and the hypertensive response is restored by adoptive transfer of T but not B lymphocytes [27]. These results were subsequently confirmed by Crowley et al., who showed that T cells are essential for the full development of AngII-dependent HT in immunodeficient scid mice [28] and by Mattson et al., who demonstrated a similar phenomenon in RAG1−/− rats [29]. In addition to this, mycophenolate immunosuppressive therapy which, inhibits T cell proliferation, reduces BP in Dahl salt-sensitive rats [30]. It is known that Ang II acts through the AT1 and AT2 receptors, both of which are present on the surface of T cells [26]. Furthermore, adoptive transfer of AT1 receptor deficient T cells into RAG1−/− animals leads to a blunted hypertensive response in Ang II-induced hypertension [27]. Infusion of Ang II increases the percentage of circulating T cells with effector phenotype (CD69+, CD25+, CCR5+) in both in vivo and in vitro studies [27, 31]. Additionally, T cells with effector phenotype accumulate in perivascular adipose tissue (PVAT) and kidneys, and affect endothelial function and vascular fibrosis [30,31,32, 33•]. Interestingly, a meta-analysis of GWAS data pointed polymorphisms in the SH2B3 gene as significant predictors of systolic and diastolic BP. This gene encodes for the lymphocyte adaptor protein, lnk [34]. Keeping with this, Saleh et al. have shown that the loss of lnk exacerbates Ang II-induced HT and its associated renal and vascular dysfunction. Moreover, lnk-deficient mice have increased T cell activation and cytokine production in comparison to wild-type animals [35]. Higher cytokine production by immunosenescent cytotoxic CD8+ T cells (CD28 null and positive for CD57) as well as augmentation of their number have been previously reported in hypertensive patients [36].
Among T cells, there is a distinct subset of regulatory cells. They express CD4 and CD25 surface markers and a forkhead transcription factor 3 (FOXP3). This unique subset possesses the capacity to suppress innate and adaptive immune responses [37]. Experimental HT is related to a decline in the number of Treg cells in in vitro and in vivo studies [38, 39]. Recent studies have shown that adoptive transfer of Treg cells reduces blood pressure [40, 41] and ameliorates endothelial function in Ang II-treated animals [39]. Further studies have shown this is accompanied by the attenuation of NADPH oxidase activity, which is critical in the development of vascular dysfunction [42, 43].
B Cells
Clinical and experimental HT is associated with raised serum IgG, IgA or IgM antibodies produced by B cells [44]. Although, transfer of B cells did not restore HT in Ang II-infused RAG1−/− mice [27], B cell activation does appear to be dependent on highly specific interactions with T cells [45] which are absent in RAG1−/− animals. Recently, Drummond’s group has shown that Ang II infusion leads to increased production of antibodies by activated B cells. Genetic deficiency of B-cell-activating factor receptor, or pharmacological depletion of B cells, protects against BP elevation and the end organ sequelae of Ang II such as collagen deposition and aortic stiffness. These effects are restored by the adoptive transfer of B cells [46].
Monocytes and Macrophages
Monocytes and macrophages have been implicated in various models of experimental HT [47, 48]. Ang II-induced HT is associated with an increased number of circulating monocytes [49, 50], and their elimination leads to decreased severity of HT, associated reduction of vascular ROS generation and improvement of vascular function [49]. Monocytes are circulating precursors of macrophages, which accumulate in the PVAT, adventitia and kidneys during HT [4, 50, 51]. Infiltrating macrophages release pro-inflammatory mediators and produce free radicals via NOX2 NADPH oxidase that changes vascular homeostasis [52,53,54]. Macrophage colony-stimulating factor (m-CSF) deficiency is associated with attenuated Ang II-induced HT, arterial remodelling, endothelial dysfunction, superoxide generation, NADPH oxidase activation and vascular inflammation [47]. Correspondingly, pharmacological blockade of macrophage CCR2 receptors, using INCB3344, prevents macrophage accumulation and reverses DOCA salt and Ang II-induced HT [50, 51].
Toll-like receptors (TLRs) have an important role in the activation of macrophages and monocytes [55]. They provoke cytokine and chemokine production through activation of NF-κB (nuclear factor kappa B) [56]. TLR4 is upregulated in Ang II-induced HT. Anti-TLR4 antibody treatment normalises BP and reduces inflammation and vascular changes associated with HT through MyD88-dependent activation and JNK/NF-κB signalling pathway [57]. Similarly, neutralization of TLR4 reduces BP and augmented vascular contractility in adult spontaneously hypertensive rats [58]. Finally, upon activation, macrophages and monocytes can activate T cells via antigen presentation, expression of costimulatory ligands and release of mediators that modulate their function and/or chemotaxis [53, 55].
Dendritic Cells
Evidence suggests that dendritic cells (DCs) play a role in the development of HT. DCs from hypertensive animals produce an increased amount of superoxide and a wide range of cytokines (IL-1β, IL-6, IL-23), which affect T cell polarization into the inflammatory phenotype [59]. Transfer of DCs from hypertensive donor mice into C57BL/6 mice results in the development of severe HT in response to sub-pressor dose of Ang II while having no effect in mice that received DCs from control animals [59]. Hypertensive mice have demonstrated higher levels of DCs with increased expression of costimulatory ligands CD80 and CD86, which are the hallmark of their activation [60]. Moreover, treatment with the pharmacological agent CTLA4-Ig (blocking B7-dependent costimulation) resulted in reduction of BP in both Ang II and DOCA salt-induced HT [60]. Chronic oxidative stress, associated with HT, leads to formation of immunogenic isoketal-protein adducts, which accumulate in DCs and promote T cell activation. Interestingly, increased isoketal adducts are also observed in immune cells of hypertensive patients [59].
Natural Killer Cells
Studies of Taherzadeh et al. have shown that NK gene complex is an important determinant to genetically determined sensitivity to develop HT and associated vascular remodelling in L-NAME-induced HT in mice [61]. An increased number of NK cells are also observed in the circulation of pregnant hypertensive rats [62]. Moreover, depletion of NK cells leads to protection from Ang II-induced vascular dysfunction [63].
Neutrophils
There are inconsistent results from studies investigating the role of neutrophils in HT. Pharmacological depletion of this subpopulation of leukocytes was associated with a significant fall in systolic BP in vivo and an attenuation in phenylephrine-induced vasoconstriction [64]. Conversely, selective depletion of circulating neutrophils protected against oxidative stress but not against the development of Ang II-induced HT [65]. Similarly, restoration of neutrophils in LysMiDTR-depleted mice with monocytes did not restore pathophysiological action of Ang II [49].
Cytokines as Key Mediators in Hypertension
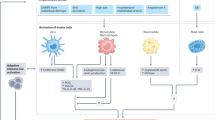
During progression of HT, immune cells accumulate in target organs, of which kidneys and the vasculature are particularly vulnerable [4, 33•, 66] (Figure 1). These cells produce potent cytokines that affect vascular and renal function, which are essential for the development of HT. In recent years, numerous cytokines with a crucial role in HT have been reported.
Tumour Necrosis Factor Alpha
Tumour necrosis factor alpha (TNF-α) is produced by many cell types including immune cells, vascular cells and adipocytes [67]. Various studies have shown that HT is associated with elevated production of TNF-α by different immune cells and a subsequent rise is observed in the circulation [27, 62, 68, 69]. Blockade of AT1 receptors in patients with HT results in a significant reduction of circulating levels of TNF-α. Mice lacking TNF-α gene or mice treated with etanercept (TNF-α antagonist) do not develop HT in response to Ang II [27, 68]. Replacement therapy with recombinant TNF-α restores action of Ang II [68]. Stimulation of endothelial cells with TNF-α decrease eNOS expression [70] by destabilisation of eNOS mRNA [71], which impairs ability of ECs to produce NO. TNF-α activates NF-κB and NADPH oxidase [70], which play an important role in the induction of oxidative stress and overexpression of both chemokines and adhesion molecules [68].
Interferon Gamma
Another pro-inflammatory cytokine produced by various immune cells which plays a role in HT is interferon gamma (IFN-γ) [63, 72]. Experimental HT is associated with increased production of IFN-γ by activated T cells and NK cells [4, 35, 63]. The knock down of IFN-γ results in a blunted increase of Ang II-induced murine BP [35]. In contrast, this phenomenon is not observed in IFN-γ receptor-1-deficient mice [73]. Loss of lnk exacerbates production of IFN-γ by CD8+ lymphocytes as well as enhances impairment of endothelial-dependent relaxation as compared to wild-type mice [35]. Interestingly, incubation of aortic segments with IFN-γ ex vivo promotes endothelial dysfunction that is partially reversed by preincubation with PEG-SOD [4], which ameliorates oxidative stress in vasculature [74]. IFN-γ also has a strong impact on superoxide production via upregulation of the expression and activity of NADPH oxidases in human aortic smooth muscle cells [75]. It acts directly on VSMC to induce their proliferation and apoptosis [76, 77]. Furthermore, neutralization of IFN-γ biologic action prevents outward vascular remodelling of human coronary arteries induced by allogenic T cells in SCID/beige mice [78]. IFN-γ affects the RAS system as well as sodium-proton-exchanger type 3 transporter in the kidneys, which leads to increased production of angiotensinogen and modulation of sodium absorption, respectively [79, 80].
Interleukin 6
Interleukin 6 (IL-6) is produced by a variety of cells, including DCs, macrophages, monocytes, T cells and vascular cells [81]. High levels of IL-6 correlate with increased BP and may be an independent risk factor for HT [81, 82]. In addition to this, the IL-6 level is reduced after treatment with Ang II-receptor blockade [83]. The increase of IL-6 is also observed in many models of experimental HT [84,85,86] strongly suggesting the essential role of IL-6 in HT. Treatment of mice with IL-6 not only increases vascular AT1 receptor expression but also induces vasoconstriction, oxidative stress and impairs endothelial function [87]. IL-6 mediates elevation of superoxide production and endothelial impairment by affecting NO-cGMP signalling pathway [88]. Furthermore, IL-6 has been reported to play an important role in VSMC migration and proliferation leading to vascular medial hypertrophy [89, 90]. Mice lacking IL-6 are protected against the action of Ang II and stress-induced HT [81, 85, 91]. Moreover, IL-6 promotes polarization of CD4+ T cells to produce IL-17 [92].
Interleukin 17
Interleukin 17 (IL-17) is produced mainly by the unique subpopulation of CD4+ cells called TH17. Additionally, production of IL-17 was reported in γ/δ cells, subsets of CD8+ T cells, some B cells and NK cells [93]. Several reports indicate that IL-17 contributes to CVD [94,95,96]. Plasma levels of IL-17 are increased in humans and animals with HT [97,98,99,100]. Administration of recombinant IL-17 in mice causes a modest elevation of BP in the absence of other hypertensive stimuli [98]. Furthermore, genetic deletion or pharmacological blockade of IL-17 protects animals against Ang II and DOCA salt-induced HT, oxidative stress and endothelial dysfunction [97, 100]. Nguyen et al. have shown that IL-17 induces phosphorylation of Thr495 on the eNOS in Rho/Rho-kinase-dependent manner leading to decrease in NO production and impairment of Ach-induced relaxation in ex vivo studies [98]. Moreover, IL-17 activates ECs as demonstrated by elevated expression of adhesion molecules and other immune cells chemoattractants [101]. Mice lacking IL-17 are protected against perivascular inflammation [100]. Recently, Harrison’s group has shown that T cells, and especially TH17 cells, play crucial role in enhanced collagen deposition in adventitia and aortic stiffening in experimental HT [72]. It is evident that Il-17 induces expression of mRNA for collagens in p38MAP-kinase-dependent fashion leading to excessive collagen deposition and loss of aortic compliance [72]. Finally, IL-17 can induce or synergize effect of other pro-inflammatory cytokines [102] leading to perturbation between pro- and anti-inflammatory factors.
Interleukin 10
In contrast to previously mentioned cytokines, interleukin 10 (IL-10) possesses anti-inflammatory properties. IL-10 not only suppresses production of TNF-α, IFN-γ and IL-6 by various immune cells [103], but also blocks the activity of pro-inflammatory transcription factors such as NF-κB [104]. It is produced by T cells, mainly Treg, DCs and macrophages [105, 106]. The IL-10 -627C/C polymorphism, associated with increased expression of IL-10, reduces an incidence of HT in Russian Tatars [107]. Hypertensive patients treated with AT1 receptor blockers or ACE inhibitors are characterized by an elevated serum IL-10 level [108]. Furthermore, there is much evidence that IL-10 blunts high BP in experimental models of HT, including preeclampsia [42, 109, 110]. Mice lacking IL-10 exhibit enhanced HT, endothelium dysfunction and increased superoxide production in response to Ang II compared with wild-type animals [63]. Similarly, incubation of the IL-10−/− vessels with Ang II reduces their relaxation and enhances superoxide production as compared to wild-type vessels ex vivo [111]. On the other hand, administration of IL-10 or an antioxidant can restore Ang II-induced endothelial dysfunction [112]. IL-10, acting on ECs, upregulates expression, activity and phosphorylation of eNOS [113] and further inhibits activation of p38 MAP-kinase, which stimulates production of pro-inflammatory cytokines and regulates NADPH oxidase activity [114, 115].
Clinical Evidence Linking Immune System and Hypertension
As stated above, experimental HT is associated with activated immune cells and their depletion/reduction very often results in normalization of BP. Based on the fact that immunosuppressive therapy is not currently clinically justified in patients with HT, we cannot countercheck these experimental observations directly. However, there is increasing evidence supporting the role of an immune component in the pathogenesis of HT in humans.
The third National Health and Nutrition Examination Survey (NHANES III) analysed data from 5626 participants and revealed a higher number of circulating leukocytes in hypertensive than in normotensive participants as well as the correlation between their number and systolic BP [116]. Increased BP is also observed in patients after infusion of allo-activated T cells during cancer treatment [117]. Conversely, HIV-infected patients have lower incidence of abnormally high BP [118]. Furthermore, immunosuppressive agents, which are not nephrotoxic, can reduce the prevalence of clinical HT [119, 120]. Reduction of BP with combination of telmisartan and rosuvastatin is related to a decrease of TH17/Treg ratio and of proinflammatory cytokines in hypertensive patients with carotid atherosclerosis [121]. Also, hereditary neutropenia, observed in Yemenites, causes small but statistically significant decline in systolic and diastolic BP in comparison to non-Yemenites population [64].
Increased BP in humans is associated with elevation of many pro-inflammatory mediators produced by immune cells such as TNF-α, IFN-γ, IL-6, IL-17 and decrease of anti-inflammatory cytokines like IL-10 [66, 69, 81, 82, 98]. This is also supported by the fact that polymorphisms of TNF-α and IL-6 are associated with human HT [122, 123].
Importantly, pro-inflammatory cytokines, mentioned above, serve not only as markers of inflammation but also confer risk for many CVD.
Other Risk Factors Affecting Development of Hypertension and Their Link with Immune System
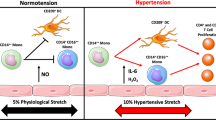
Physiological stress is a significant factor for many CVD [124]. Moreover, sympathetic nervous system participates in development of HT [11, 12]. Marvar et al. have demonstrated that repeated daily stress elevates BP in mice. This was associated with activation of the immune system i.e. increase of circulating T cells expressing CD69 and CD44high markers. Moreover, Ang II-infused mice exposed to chronic stress displayed greater BP than non-stressed animals. In contrast, repeated stress did not affect BP in T and B cells depleted RAG1−/− mice [125].
Another risk factor for HT is obesity, which is also associated with chronic inflammation, vascular remodelling and endothelial dysfunction [126, 127]. Cells of both the innate and adoptive immune system reside in adipose tissue and participate in these pathophysiological changes [128]. Moreover, immune cells produce cytokines that can modulate expression of classical adipokines. Production of anti-inflammatory adiponectin can be inhibited by pro-inflammatory mediators such as TNF-α, IL-6 and IL-17. At the same time, pro-inflammatory cytokines can induce production of leptin which is critical in the development of obesity [33•].
Some studies have shown an association between air pollution exposure and the risk of HT [129]. Exposure to PM2.5 (particulate matter <2.5 μm) and ozone increases blood pressure in humans [130]. Similarly, PM2.5 exposure in mice induces endothelial dysfunction and mild elevation in BP, which is associated with increased levels of inflammatory cytokines in blood (TNF-α, IL-6). In addition to this, there is also an increased number of activated macrophages in adipose tissue and increased vascular adhesion of monocytes [131]. Finally, pollutant-induced oxidative stress may affect vascular function [132].
Micro-RNA as Novel Immune Players in Hypertension
Micro-RNAs (miRNAs) are implicated in the intricate control of genes throughout the body; therefore, it is understandable that they can be associated with immune mechanisms of hypertension (Fig. 1). MiRNAs are 18–22 nucleotides in length and do not code for a gene but may affect expression of other target genes. This is achieved through a negative regulation via binding of its complex (the RNA-induced silencing complex) to the 3′ untranslated region (3′ UTR) of its target messenger RNA (mRNA). Sixty percent of protein coding genes have miRNA target sites in their 3′ UTR, and a single miRNA may impact the expression of multiple mRNAs. MiRNAs are involved in both the cellular and clinical manifestations of various CVD diseases such as HT [133], coronary artery disease [134] and cardiac hypertrophy with subsequent heart failure [135]. Further to this, miRNAs are implicated in the immune system in the context of chronic immune disorders [136,137,138,139]. Two miRNAs are discussed, which are considered to be pro-inflammatory in nature and demonstrated associations with chronic inflammatory arthritides as well as with cardiovascular manifestations of HT.

Immune mechanism of hypertension
miR-21
Micro-RNA 21 is an extensively investigated miR in the context of the cardiovascular system [140•]. MiR-21 is elevated in peripheral blood mononuclear cells (PBMCs) of hypertensive patients and is associated with the degree of LVH detected in this group [141]. In coronary artery disease, McDonald et al. demonstrated that eliminating miR-21 reduces in stent restenosis via anti-inflammatory effects of M2 macrophages, lowering number of CD3+ T cells and a reduction of Ly6c+cells level [134] (which differentiate to become the pro-inflammatory M1 phenotype). It is widely accepted that nitric oxide synthase is a marker for M1 macrophages and subsequently M1 cells produce ROS. This response was likely evolved to kill pathogens in the micro environment. That being said, it is now known that ROS elevate BP and activate pro-fibrotic signalling pathways as reviewed in many excellent reviews including this piece by Montezano and Touyz [142]. It is noteworthy that iNOS is not expressed by macrophages in humans; however, a clinical study has shown a relationship between elevated miR-21 and endothelial dysfunction with an associated reduction in NO and eNOS levels in hypertensive patients [143]. miR-21 can modulate T cell responses including exerting influence on cytokine production [144]. Not only miR-21 is induced by T cell activation and enhances the T cell immune response [145] but also miR-21 has been shown to induce proliferation of CD4+ T cells in murine models of systemic lupus erythematosus (SLE) and in patients with SLE [146, 147]. Relating to the production of IL-17 from CD4+ cells, previously mentioned in this work, this could link miR-21 to the increased cardiovascular risk observed in patients with lupus and other inflammatory arthritides. Further to this, miR-21 could serve as a biomarker for CVD in this patient group. miR-21 is a positive regulator of the expression of the transcription factor, FOXP3 [148]. As previously mentioned, Treg cells express FOXP3 and there is an association between a lower number of Treg cells and HT [38, 39]. This conflicts with the previously understood concepts that miR-21 is associated with pathological processes seen in elevated BP suggesting the full mechanism is not yet known, thus bringing together the development of HT, inflammatory cell lines, the coronary arterial disease sequelae of HT and miR-21.
miR-155
The endothelium is the single-cell surface layer lining the entire vascular tree, and inflammation, the response to injury, is observed in the endothelium in HT. The endothelium is the source of eNOS, which underpins much of the vascular sequelae of HT. miR-155 reduces eNOS expression [149]. Mir-155 is regarded as a pro-inflammatory miR and is necessary for normal B cell and T cell function. In miR-155 KO mice, it has been shown that it is implicated in the regulation of many genes including chemokines and cytokines [150, 151]. Further to this, TNF-α increases miR-155 expression with subsequent negative regulation of eNOS [133, 152]. TNF-α has also been shown to increase miR-155 expression in macrophages and monocytes during an inflammatory response [153] helping to explain this phenomenon in HT. In short, the effects of TNF-α discussed earlier in this paper are closely mediated by miR-155. MiR-155 has been shown to regulate the AT1 receptor in rat cardiomyocytes [154], rat aortic adventitial-derived fibroblasts [155] and in human PBMCs where it was also found to correlate with BP [151]. Interestingly, rare allele of rs5186 polymorphism, located in the AT1 receptor, blocks adhesion of mir-155 to the AT1 receptor 3′ UTR [156]. This observation may explain that rs5186 was a risk factor for HT in several epidemiological studies, although overall, the clinical effect of this polymorphism is unclear and further research, taking into account different subpopulations of patients with HT, are needed to verify its role in HT [157]. MiR-155 level in aortic tissue of adult spontaneously hypertensive rats negatively correlates with the blood pressure [158]. This has translated into a human study that examined miR levels in PBMCs and demonstrated lower circulating pro-inflammatory cytokines including TNF-α in patients who had downregulation of miR-155 [159]. It has been shown that the 5p strand of miR-155 is upregulated in fibroblasts in the synovial fluid taken from patients with rheumatoid arthritis [160], suggesting that miR-155 plays an important role in a chronic inflammatory response. In summary, miR-155 is another miR, which provides an association between inflammatory arthritis and HT. Moreover, miR-155 is associated with hypertension and is implicated in a pro-inflammatory phenotype. It is likely that these effects may have an impact in either HT onset or severity.
Conclusions
In summary, hypertension is associated with significant activation of immune and inflammatory systems and shares several functional differences with other immune-mediated diseases. Low-grade inflammation is prominent and further understanding of specific cytokine and chemokine milieu, similarities and differences between hypertension pathogenesis and atherosclerosis will shed a light on possible new therapeutic strategies to limit vascular and renal complications of HT. Indeed, in experimental models, immune-targeted therapies prevent vascular and renal damage and can alleviate hypertension. Evidence in humans is now urgently needed.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17(11):1402–9. doi:10.1038/nm.2541.
Wang Y, Wang QJ. The prevalence of prehypertension and hypertension among US adults according to the new joint national committee guidelines: new challenges of the old problem. Arch Intern Med. 2004;164(19):2126–34.
Fields LE, Burt VL, Cutler JA, Hughes J, Roccella EJ, Sorlie P. The burden of adult hypertension in the United States 1999 to 2000: a rising tide. Hypertension. 2004;44(4):398–404.
Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D, et al. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J: Off Publ Fed Am Soc Exp Biol. 2016;30(5):1987–99. doi:10.1096/fj.201500088R.
Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, et al. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res. 2014;114(4):616–25. doi:10.1161/CIRCRESAHA.114.302157.
Montezano AC, Nguyen Dinh Cat A, Rios FJ, Touyz RM. Angiotensin II and vascular injury. Curr Hypertens Rep. 2014;16(6):431. doi:10.1007/s11906-014-0431-2.
Wilk G, Osmenda G, Matusik P, Nowakowski D, Jasiewicz-Honkisz B, Ignacak A, et al. Endothelial function assessment in atherosclerosis: comparison of brachial artery flowmediated vasodilation and peripheral arterial tonometry. Pol Arch Med Wewn. 2013;123(9):443–52.
Dahl LK, Heine M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res. 1975;36(6):692–6.
Curtis JJ, Luke RG, Dustan HP, Kashgarian M, Whelchel JD, Jones P, et al. Remission of essential hypertension after renal transplantation. N Engl J Med. 1983;309(17):1009–15. doi:10.1056/NEJM198310273091702.
Grassi G. Assessment of sympathetic cardiovascular drive in human hypertension: achievements and perspectives. Hypertension. 2009;54(4):690–7. doi:10.1161/HYPERTENSIONAHA.108.119883.
Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, et al. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55(2):277–83. doi:10.1161/HYPERTENSIONAHA.109.142646. 6p following 83.
Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension. 2013;61(2):382–7. doi:10.1161/HYPERTENSIONAHA.111.00546.
•• Matusik P, Guzik B, Weber C, Guzik TJ. Do we know enough about the immune pathogenesis of acute coronary syndromes to improve clinical practice? Thromb Haemost. 2012;108(3):443–56. doi:10.1160/TH12-05-0341. This paper provides a comprehensive review of the role of the immune system and inflammation in ACS.
Skiba DS, Nosalski R, Mikolajczyk TP, Siedlinski M, Rios FJ, Montezano AC, et al. Antiatherosclerotic effect of Ang- (1–7) non-peptide mimetic (AVE 0991) is mediated by inhibition of perivascular and plaque inflammation in early atherosclerosis. Br J Pharmacol. 2016. doi:10.1111/bph.13685.
Caillon A, Schiffrin EL. Role of inflammation and immunity in hypertension: recent epidemiological, laboratory, and clinical evidence. Curr Hypertens Rep. 2016;18(3):21. doi:10.1007/s11906-016-0628-7.
Devaraj S, Siegel D, Jialal I. Statin therapy in metabolic syndrome and hypertension post-JUPITER: what is the value of CRP? Curr Atheroscler Rep. 2011;13(1):31–42. doi:10.1007/s11883-010-0143-2.
Vongpatanasin W, Thomas GD, Schwartz R, Cassis LA, Osborne-Lawrence S, Hahner L, et al. C-reactive protein causes downregulation of vascular angiotensin subtype 2 receptors and systolic hypertension in mice. Circulation. 2007;115(8):1020–8. doi:10.1161/CIRCULATIONAHA.106.664854.
Fichtlscherer S, Rosenberger G, Walter DH, Breuer S, Dimmeler S, Zeiher AM. Elevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery disease. Circulation. 2000;102(9):1000–6.
Qamirani E, Ren Y, Kuo L, Hein TW. C-reactive protein inhibits endothelium-dependent NO-mediated dilation in coronary arterioles by activating p38 kinase and NAD(P)H oxidase. Arterioscler Thromb Vasc Biol. 2005;25(5):995–1001. doi:10.1161/01.ATV.0000159890.10526.1e.
Singh U, Devaraj S, Vasquez-Vivar J, Jialal I. C-reactive protein decreases endothelial nitric oxide synthase activity via uncoupling. J Mol Cell Cardiol. 2007;43(6):780–91. doi:10.1016/j.yjmcc.2007.08.015.
Kobayashi S, Inoue N, Ohashi Y, Terashima M, Matsui K, Mori T, et al. Interaction of oxidative stress and inflammatory response in coronary plaque instability: important role of C-reactive protein. Arterioscler Thromb Vasc Biol. 2003;23(8):1398–404. doi:10.1161/01.ATV.0000081637.36475.BC.
White FN, Grollman A. Autoimmune factors associated with infraction of kidney. Nephron. 1964;204:93–102.
Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med. 1967;25(2):257–64.
Olsen F. Transfer of arterial hypertension by splenic cells from DOCA-salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Scand C Immuno. 1980;88(1):1–5.
Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand C Immuno. 1976;84(6):523–8.
Zhang J, Crowley SD. The role of type 1 angiotensin receptors on T lymphocytes in cardiovascular and renal diseases. Curr Hypertens Rep. 2013;15(1):39–46. doi:10.1007/s11906-012-0318-z.
Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–60. doi:10.1084/jem.20070657.
Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298(4):R1089–97. doi:10.1152/ajpregu.00373.2009.
Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304(6):R407–14. doi:10.1152/ajpregu.00304.2012.
De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2010;298(4):R1136–42. doi:10.1152/ajpregu.00298.2009.
Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, et al. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2009;296(2):R208–16. doi:10.1152/ajpregu.90521.2008.
Filip M, Maciag J, Nosalski R, Korbut R, Guzik T. Endothelial dysfunction related to oxidative stress and inflammation in perivascular adipose tissue. Postepy Biochem. 2012;58(2):186–94.
• Nosalski R, Guzik TJ. Perivascular adipose tissue inflammation in vascular disease. British journal of pharmacology. 2017. doi:10.1111/bph.13705. This review emphasizing the role of the immune system in perivascular inflammation which is associated with many cardiovascular diseases including hypertension.
Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41(6):677–87. doi:10.1038/ng.384.
Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, et al. Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest. 2015;125(3):1189–202. doi:10.1172/JCI76327.
Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, et al. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. 2013;62(1):126–33. doi:10.1161/HYPERTENSIONAHA.113.00689.
Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8(7):523–32. doi:10.1038/nri2343.
Viel EC, Lemarie CA, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol. 2010;298(3):H938–44. doi:10.1152/ajpheart.00707.2009.
Matrougui K, Abd Elmageed Z, Kassan M, Choi S, Nair D, Gonzalez-Villalobos RA, et al. Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol. 2011;178(1):434–41. doi:10.1016/j.ajpath.2010.11.034.
Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, et al. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012;59(2):324–30. doi:10.1161/HYPERTENSIONAHA.111.181123.
Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57(3):469–76. doi:10.1161/HYPERTENSIONAHA.110.162941.
Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2534–42. doi:10.1161/ATVBAHA.111.233262.
Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab. 2014;25(9):452–63. doi:10.1016/j.tem.2014.06.012.
Chan CT, Lieu M, Toh BH, Kyaw TS, Bobik A, Sobey CG, et al. Antibodies in the pathogenesis of hypertension. Biomed Res Int. 2014;2014:504045. doi:10.1155/2014/504045.
Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–60. doi:10.1146/annurev.iy.11.040193.001555.
Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, et al. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension. 2015;66(5):1023–33. doi:10.1161/HYPERTENSIONAHA.115.05779.
De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25(10):2106–13. doi:10.1161/01.atv.0000181743.28028.57.
Franco M, Martinez F, Quiroz Y, Galicia O, Bautista R, Johnson RJ, et al. Renal angiotensin II concentration and interstitial infiltration of immune cells are correlated with blood pressure levels in salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2007;293(1):R251–6. doi:10.1152/ajpregu.00645.2006.
Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124(12):1370–81. doi:10.1161/CIRCULATIONAHA.111.034470.
Moore JP, Vinh A, Tuck KL, Sakkal S, Krishnan SM, Chan CT, et al. M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am J Physiol Heart Circ Physiol. 2015;309(5):H906–17. doi:10.1152/ajpheart.00821.2014.
Chan CT, Moore JP, Budzyn K, Guida E, Diep H, Vinh A, et al. Reversal of vascular macrophage accumulation and hypertension by a CCR2 antagonist in deoxycorticosterone/salt-treated mice. Hypertension. 2012;60(5):1207–12. doi:10.1161/HYPERTENSIONAHA.112.201251.
Kotsias F, Hoffmann E, Amigorena S, Savina A. Reactive oxygen species production in the phagosome: impact on antigen presentation in dendritic cells. Antioxid Redox Signal. 2013;18(6):714–29. doi:10.1089/ars.2012.4557.
Shirai T, Hilhorst M, Harrison DG, Goronzy JJ, Weyand CM. Macrophages in vascular inflammation—from atherosclerosis to vasculitis. Autoimmunity. 2015;48(3):139–51. doi:10.3109/08916934.2015.1027815.
Harrison DG, Guzik TJ. Macrophages come to mind as keys to cognitive decline. J Clin Invest. 2016;126(12):4393–5. doi:10.1172/jci91277.
Weber C, Shantsila E, Hristov M, Caligiuri G, Guzik T, Heine GH, et al. Role and analysis of monocyte subsets in cardiovascular disease. Joint consensus document of the European Society of Cardiology (ESC) Working Groups “Atherosclerosis & Vascular Biology” and “Thrombosis”. Thromb Haemost. 2016;116(4):626–37. doi:10.1160/TH16-02-0091.
Singh MV, Abboud FM. Toll-like receptors and hypertension. Am J Physiol Regul Integr Comp Physiol. 2014;307(5):R501–4. doi:10.1152/ajpregu.00194.2014.
Hernanz R, Martinez-Revelles S, Palacios R, Martin A, Cachofeiro V, Aguado A, et al. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br J Pharmacol. 2015;172(12):3159–76. doi:10.1111/bph.13117.
Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, et al. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond). 2012;122(11):535–43. doi:10.1042/CS20110523.
Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124(10):4642–56. doi:10.1172/JCI74084.
Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122(24):2529–37. doi:10.1161/circulationaha.109.930446.
Taherzadeh Z, VanBavel E, de Vos J, Matlung HL, van Montfrans G, Brewster LM, et al. Strain-dependent susceptibility for hypertension in mice resides in the natural killer gene complex. Am J Physiol Heart Circ Physiol. 2010;298(4):H1273–82. doi:10.1152/ajpheart.00508.2009.
Small HY, Nosalski R, Morgan H, Beattie E, Guzik TJ, Graham D, et al. Role of tumor necrosis factor-alpha and natural killer cells in uterine artery function and pregnancy outcome in the stroke-prone spontaneously hypertensive rat. Hypertension. 2016;68(5):1298–307. doi:10.1161/HYPERTENSIONAHA.116.07933.
Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M, et al. Angiotensin II-induced vascular dysfunction depends on interferon-gamma-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol. 2013;33(6):1313–9. doi:10.1161/ATVBAHA.113.301437.
Morton J, Coles B, Wright K, Gallimore A, Morrow JD, Terry ES, et al. Circulating neutrophils maintain physiological blood pressure by suppressing bacteria and IFNgamma-dependent iNOS expression in the vasculature of healthy mice. Blood. 2008;111(10):5187–94. doi:10.1182/blood-2007-10-117283.
Yildirim A, Russell J, Yan LS, Senchenkova EY, Granger DN. Leukocyte-dependent responses of the microvasculature to chronic angiotensin II exposure. Hypertension. 2012;60(6):1503–9. doi:10.1161/HYPERTENSIONAHA.112.198465.
Itani HA, McMaster Jr WG, Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, et al. Activation of human T cells in hypertension: studies of humanized mice and hypertensive humans. Hypertension. 2016;68(1):123–32. doi:10.1161/HYPERTENSIONAHA.116.07237.
Mendizabal Y, Llorens S, Nava E. Hypertension in metabolic syndrome: vascular pathophysiology. Int J Hypertens. 2013;2013:230868. doi:10.1155/2013/230868.
Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51(5):1345–51. doi:10.1161/HYPERTENSIONAHA.107.102152.
Manabe S, Okura T, Watanabe S, Fukuoka T, Higaki J. Effects of angiotensin II receptor blockade with valsartan on pro-inflammatory cytokines in patients with essential hypertension. J Cardiovasc Pharmacol. 2005;46(6):735–9.
Hot A, Lenief V, Miossec P. Combination of IL-17 and TNFalpha induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann Rheum Dis. 2012;71(5):768–76. doi:10.1136/annrheumdis-2011-200468.
Neumann P, Gertzberg N, Johnson A. TNF-alpha induces a decrease in eNOS promoter activity. Am J Physiol Lung Cell Mol Physiol. 2004;286(2):L452–9. doi:10.1152/ajplung.00378.2002.
Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126(1):50–67. doi:10.1172/JCI80761.
Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, et al. Interferon-gamma signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60(6):1430–6. doi:10.1161/HYPERTENSIONAHA.112.199265.
Guzik TJ, Olszanecki R, Sadowski J, Kapelak B, Rudzinski P, Jopek A, et al. Superoxide dismutase activity and expression in human venous and arterial bypass graft vessels. J Physiol Pharmacol: Off J Pol Physiol Soc. 2005;56(2):313–23.
Manea SA, Todirita A, Raicu M, Manea A. C/EBP transcription factors regulate NADPH oxidase in human aortic smooth muscle cells. J Cell Mol Med. 2014;18(7):1467–77. doi:10.1111/jcmm.12289.
Wang Y, Bai Y, Qin L, Zhang P, Yi T, Teesdale SA, et al. Interferon-gamma induces human vascular smooth muscle cell proliferation and intimal expansion by phosphatidylinositol 3-kinase dependent mammalian target of rapamycin raptor complex 1 activation. Circ Res. 2007;101(6):560–9. doi:10.1161/CIRCRESAHA.107.151068.
Rosner D, Stoneman V, Littlewood T, McCarthy N, Figg N, Wang Y, et al. Interferon-gamma induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K- and Akt-dependent mechanism. Am J Pathol. 2006;168(6):2054–63. doi:10.2353/ajpath.2006.050473.
Wang Y, Burns WR, Tang PC, Yi T, Schechner JS, Zerwes HG, et al. Interferon-gamma plays a nonredundant role in mediating T cell-dependent outward vascular remodeling of allogeneic human coronary arteries. FASEB J: Off Publ Fed Am Soc Exp Biol. 2004;18(3):606–8. doi:10.1096/fj.03-0840fje.
Satou R, Miyata K, Gonzalez-Villalobos RA, Ingelfinger JR, Navar LG, Kobori H. Interferon-gamma biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J: Off Publ Fed Am Soc Exp Biol. 2012;26(5):1821–30. doi:10.1096/fj.11-195198.
Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, et al. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-gamma−/− and interleukin-17A−/− mice. Hypertension. 2015;65(3):569–76. doi:10.1161/HYPERTENSIONAHA.114.04975.
Lee DL, Leite R, Fleming C, Pollock JS, Webb RC, Brands MW. Hypertensive response to acute stress is attenuated in interleukin-6 knockout mice. Hypertension. 2004;44(3):259–63. doi:10.1161/01.HYP.0000139913.56461.fb.
Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101(15):1767–72.
Vazquez-Oliva G, Fernandez-Real JM, Zamora A, Vilaseca M, Badimon L. Lowering of blood pressure leads to decreased circulating interleukin-6 in hypertensive subjects. J Hum Hypertens. 2005;19(6):457–62. doi:10.1038/sj.jhh.1001845.
Lamarca B, Brewer J, Wallace K. IL-6-induced pathophysiology during pre-eclampsia: potential therapeutic role for magnesium sulfate? Int J Interferon Cytokine Mediat Res. 2011;2011(3):59–64. doi:10.2147/IJICMR.S16320.
Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension. 2010;56(5):879–84. doi:10.1161/HYPERTENSIONAHA.110.158071.
Luther JM, Gainer JV, Murphey LJ, Yu C, Vaughan DE, Morrow JD, et al. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension. 2006;48(6):1050–7. doi:10.1161/01.HYP.0000248135.97380.76.
Wassmann S, Stumpf M, Strehlow K, Schmid A, Schieffer B, Bohm M, et al. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res. 2004;94(4):534–41. doi:10.1161/01.RES.0000115557.25127.8D.
Orshal JM, Khalil RA. Interleukin-6 impairs endothelium-dependent NO-cGMP-mediated relaxation and enhances contraction in systemic vessels of pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2004;286(6):R1013–23. doi:10.1152/ajpregu.00729.2003.
Chava KR, Karpurapu M, Wang D, Bhanoori M, Kundumani-Sridharan V, Zhang Q, et al. CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2009;29(6):809–15. doi:10.1161/ATVBAHA.109.185777.
Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007;27(12):2576–81. doi:10.1161/ATVBAHA.107.153080.
Coles B, Fielding CA, Rose-John S, Scheller J, Jones SA, O’Donnell VB. Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am J Pathol. 2007;171(1):315–25. doi:10.2353/ajpath.2007.061078.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–8. doi:10.1038/nature04753.
Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res. 2014;59(1–3):243–53. doi:10.1007/s12026-014-8548-6.
Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE, et al. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31(7):1565–72. doi:10.1161/ATVBAHA.111.227629.
Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102(2):257–64. doi:10.1161/CIRCRESAHA.107.158220.
Behnamfar N, Zibaeenezhad MJ, Golmoghaddam H, Doroudchi M. CD45RO+ memory T-cells produce IL-17 in patients with atherosclerosis. Cell Mol Biol (Noisy-le-Grand). 2015;61(8):17–23.
Amador CA, Barrientos V, Pena J, Herrada AA, Gonzalez M, Valdes S, et al. Spironolactone decreases DOCA-salt-induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension. 2014;63(4):797–803. doi:10.1161/HYPERTENSIONAHA.113.02883.
Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97(4):696–704. doi:10.1093/cvr/cvs422.
Karbach S, Croxford AL, Oelze M, Schuler R, Minwegen D, Wegner J, et al. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler Thromb Vasc Biol. 2014;34(12):2658–68. doi:10.1161/ATVBAHA.114.304108.
Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–7. doi:10.1161/HYPERTENSIONAHA.109.145094.
Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R, et al. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol. 2010;184(8):4531–7. doi:10.4049/jimmunol.0903162.
Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL, et al. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279(4):2559–67. doi:10.1074/jbc.M308809200.
Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: a complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med. 2002;30(1 Supp):S58–63.
Hovsepian E, Penas F, Siffo S, Mirkin GA, Goren NB. IL-10 inhibits the NF-kappaB and ERK/MAPK-mediated production of pro-inflammatory mediators by up-regulation of SOCS-3 in Trypanosoma cruzi-infected cardiomyocytes. PLoS One. 2013;8(11):e79445. doi:10.1371/journal.pone.0079445.
Krause P, Morris V, Greenbaum JA, Park Y, Bjoerheden U, Mikulski Z, et al. IL-10-producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL-23 synthesis. Nat Commun. 2015;6:7055. doi:10.1038/ncomms8055.
Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10(3):170–81. doi:10.1038/nri2711.
Timasheva YR, Nasibullin TR, Zakirova AN, Mustafina OE. Association of interleukin-6, interleukin-12, and interleukin-10 gene polymorphisms with essential hypertension in Tatars from Russia. Biochem Genet. 2008;46(1–2):64–74. doi:10.1007/s10528-007-9130-x.
Schieffer B, Bunte C, Witte J, Hoeper K, Boger RH, Schwedhelm E, et al. Comparative effects of AT1-antagonism and angiotensin-converting enzyme inhibition on markers of inflammation and platelet aggregation in patients with coronary artery disease. J Am Coll Cardiol. 2004;44(2):362–8. doi:10.1016/j.jacc.2004.03.065.
Tinsley JH, South S, Chiasson VL, Mitchell BM. Interleukin-10 reduces inflammation, endothelial dysfunction, and blood pressure in hypertensive pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2010;298(3):R713–9. doi:10.1152/ajpregu.00712.2009.
Chatterjee P, Chiasson VL, Kopriva SE, Young KJ, Chatterjee V, Jones KA, et al. Interleukin 10 deficiency exacerbates toll-like receptor 3-induced preeclampsia-like symptoms in mice. Hypertension. 2011;58(3):489–96. doi:10.1161/HYPERTENSIONAHA.111.172114.
Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension. 2009;54(3):619–24. doi:10.1161/HYPERTENSIONAHA.109.137158.
Zemse SM, Hilgers RH, Webb RC. Interleukin-10 counteracts impaired endothelium-dependent relaxation induced by ANG II in murine aortic rings. Am J Physiol Heart Circ Physiol. 2007;292(6):H3103–8. doi:10.1152/ajpheart.00456.2006.
Cattaruzza M, Slodowski W, Stojakovic M, Krzesz R, Hecker M. Interleukin-10 induction of nitric-oxide synthase expression attenuates CD40-mediated interleukin-12 synthesis in human endothelial cells. J Biol Chem. 2003;278(39):37874–80. doi:10.1074/jbc.M301670200.
Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH oxidases in vascular pathology. Antioxid Redox Signal. 2014;20(17):2794–814. doi:10.1089/ars.2013.5607.
Kontoyiannis D, Kotlyarov A, Carballo E, Alexopoulou L, Blackshear PJ, Gaestel M, et al. Interleukin-10 targets p38 MAPK to modulate ARE-dependent TNF mRNA translation and limit intestinal pathology. EMBO J. 2001;20(14):3760–70. doi:10.1093/emboj/20.14.3760.
Park CS, Kim H-Y, Park H-J, Jang S-W, Ihm S-H, Lee J-M, et al. Association between the JNC 7 classification of the stages of systolic hypertension and inflammatory cardiovascular risk factors. Korean Circ J. 2007;37(12):623. doi:10.4070/kcj.2007.37.12.623.
Balsari A, Marolda R, Gambacorti-Passerini C, Sciorelli G, Tona G, Cosulich E, et al. Systemic administration of autologous, alloactivated helper-enriched lymphocytes to patients with metastatic melanoma of the lung. A phase I study. Cancer Immunol Immunother. 1986;21(2):148–55.
Palacios R, Santos J, García A, Castells E, González M, Ruiz J, et al. Impact of HAART on blood pressure in HIV-infected patients. A prospective study in a cohort of naive patients. HIV Med. 2006;7(1):10–5.
Morales JM. Influence of the new immunosuppressive combinations on arterial hypertension after renal transplantation. Kidney Int Suppl. 2002;82:S81–7. doi:10.1046/j.1523-1755.62.s82.16.x.
Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol. 2006;17(12 Suppl 3):S218–25. doi:10.1681/ASN.2006080918.
Liu Z, Zhao Y, Wei F, Ye L, Lu F, Zhang H, et al. Treatment with telmisartan/rosuvastatin combination has a beneficial synergistic effect on ameliorating Th17/Treg functional imbalance in hypertensive patients with carotid atherosclerosis. Atherosclerosis. 2014;233(1):291–9. doi:10.1016/j.atherosclerosis.2013.12.004.
Cheung BM, Ong KL, Tso AW, Leung RY, Cherny SS, Sham PC, et al. Relationship of plasma interleukin-6 and its genetic variants with hypertension in Hong Kong Chinese. Am J Hypertens. 2011;24(12):1331–7. doi:10.1038/ajh.2011.141.
Li YY. Tumor necrosis factor-alpha g308alpha gene polymorphism and essential hypertension: a meta-analysis involving 2244 participants. PLoS One. 2012;7(4):e35408. doi:10.1371/journal.pone.0035408.
Gasperin D, Netuveli G, Dias-da-Costa JS, Pattussi MP. Effect of psychological stress on blood pressure increase: a meta-analysis of cohort studies. Cad Saude Publica. 2009;25(4):715–26.
Marvar PJ, Vinh A, Thabet S, Lob HE, Geem D, Ressler KJ, et al. T lymphocytes and vascular inflammation contribute to stress-dependent hypertension. Biol Psychiatry. 2012;71(9):774–82. doi:10.1016/j.biopsych.2012.01.017.
Aghamohammadzadeh R, Unwin RD, Greenstein AS, Heagerty AM. Effects of obesity on perivascular adipose tissue vasorelaxant function: nitric oxide, inflammation and elevated systemic blood pressure. J Vasc Res. 2015;52(5):299–305. doi:10.1159/000443885.
Dinh Cat AN, Friederich-Persson M, White A, Touyz RM. Adipocytes, aldosterone and obesity-related hypertension. J Mol Endocrinol. 2016;57(1):F7–F21. doi:10.1530/JME-16-0025.
McLaughlin T, Ackerman SE, Shen L, Engleman E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest. 2017;127(1):5–13. doi:10.1172/jci88876.
Cai Y, Zhang B, Ke W, Feng B, Lin H, Xiao J, et al. Associations of short-term and long-term exposure to ambient air pollutants with hypertension: a systematic review and meta-analysis. Hypertension. 2016;68(1):62–70. doi:10.1161/HYPERTENSIONAHA.116.07218.
Urch B, Silverman F, Corey P, Brook JR, Lukic KZ, Rajagopalan S, et al. Acute blood pressure responses in healthy adults during controlled air pollution exposures. Environ Health Perspect. 2005;113(8):1052–5.
Sun Q, Yue P, Deiuliis JA, Lumeng CN, Kampfrath T, Mikolaj MB, et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation. 2009;119(4):538–46. doi:10.1161/CIRCULATIONAHA.108.799015.
Brook RD. Cardiovascular effects of air pollution. Clin Sci (Lond). 2008;115(6):175–87. doi:10.1042/CS20070444.
Sun HX, Zeng DY, Li RT, Pang RP, Yang H, Hu YL, et al. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension. 2012;60(6):1407–14. doi:10.1161/HYPERTENSIONAHA.112.197301.
McDonald RA, Halliday CA, Miller AM, Diver LA, Dakin RS, Montgomery J, et al. Reducing in-stent restenosis: therapeutic manipulation of miRNA in vascular remodeling and inflammation. J Am Coll Cardiol. 2015;65(21):2314–27. doi:10.1016/j.jacc.2015.03.549.
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456(7224):980–4. doi:10.1038/nature07511.
Chen L, Al-Mossawi MH, Ridley A, Sekine T, Hammitzsch A, de Wit J, et al. miR-10b-5p is a novel Th17 regulator present in Th17 cells from ankylosing spondylitis. Ann Rheum Dis. 2016. doi:10.1136/annrheumdis-2016-210175.
Yang Y, Wang Y, Liang Q, Yao L, Gu S, Bai X. MiR-338-5p promotes inflammatory response of fibroblast-like synoviocytes in rheumatoid arthritis via targeting SPRY1. J Cell Biochem. 2017. doi:10.1002/jcb.25883.
Alivernini S, Kurowska-Stolarska M, Tolusso B, Benvenuto R, Elmesmari A, Canestri S, et al. MicroRNA-155 influences B-cell function through PU.1 in rheumatoid arthritis. Nat Commun. 2016;7:12970. doi:10.1038/ncomms12970.
Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci U S A. 2011;108(27):11193–8. doi:10.1073/pnas.1019536108.
• Jones Buie JN, Goodwin AJ, Cook JA, Halushka PV, Fan H. The role of miRNAs in cardiovascular disease risk factors. Atherosclerosis. 2016;254:271–81. doi:10.1016/j.atherosclerosis.2016.09.067. This paper reviews the potential role of miRNAs in cardiovascular disease risk factors as well as describes their beneficial or deleterious effects.
Kontaraki JE, Marketou ME, Parthenakis FI, Maragkoudakis S, Zacharis EA, Petousis S, et al. Hypertrophic and antihypertrophic microRNA levels in peripheral blood mononuclear cells and their relationship to left ventricular hypertrophy in patients with essential hypertension. J Am Soc Hypertens. 2015;9(10):802–10. doi:10.1016/j.jash.2015.07.013.
Montezano AC, Touyz RM. Oxidative stress, Noxs, and hypertension: experimental evidence and clinical controversies. Ann Med. 2012;44 Suppl 1:S2–16. doi:10.3109/07853890.2011.653393.
Cengiz M, Yavuzer S, Kilickiran Avci B, Yuruyen M, Yavuzer H, Dikici SA, et al. Circulating miR-21 and eNOS in subclinical atherosclerosis in patients with hypertension. Clin Exp Hypertens. 2015;37(8):643–9. doi:10.3109/10641963.2015.1036064.
Wang L, He L, Zhang R, Liu X, Ren Y, Liu Z, et al. Regulation of T lymphocyte activation by microRNA-21. Mol Immunol. 2014;59(2):163–71. doi:10.1016/j.molimm.2014.02.004.
Smigielska-Czepiel K, van den Berg A, Jellema P, Slezak-Prochazka I, Maat H, van den Bos H, et al. Dual role of miR-21 in CD4+ T-cells: activation-induced miR-21 supports survival of memory T-cells and regulates CCR7 expression in naive T-cells. PLoS One. 2013;8(10):e76217. doi:10.1371/journal.pone.0076217.
Garchow BG, Bartulos Encinas O, Leung YT, Tsao PY, Eisenberg RA, Caricchio R, et al. Silencing of microRNA-21 in vivo ameliorates autoimmune splenomegaly in lupus mice. EMBO Mol Med. 2011;3(10):605–15. doi:10.1002/emmm.201100171.
Stagakis E, Bertsias G, Verginis P, Nakou M, Hatziapostolou M, Kritikos H, et al. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis. 2011;70(8):1496–506. doi:10.1136/ard.2010.139857.
Rouas R, Fayyad-Kazan H, El Zein N, Lewalle P, Rothe F, Simion A, et al. Human natural Treg microRNA signature: role of microRNA-31 and microRNA-21 in FOXP3 expression. Eur J Immunol. 2009;39(6):1608–18. doi:10.1002/eji.200838509.
Zhang J, Zhao F, Yu X, Lu X, Zheng G. MicroRNA-155 modulates the proliferation of vascular smooth muscle cells by targeting endothelial nitric oxide synthase. Int J Mol Med. 2015;35(6):1708–14. doi:10.3892/ijmm.2015.2181.
Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316(5824):608–11. doi:10.1126/science.1139253.
Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):604–8. doi:10.1126/science.1141229.
Lee KS, Kim J, Kwak SN, Lee KS, Lee DK, Ha KS, et al. Functional role of NF-kappaB in expression of human endothelial nitric oxide synthase. Biochem Biophys Res Commun. 2014;448(1):101–7. doi:10.1016/j.bbrc.2014.04.079.
O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104(5):1604–9. doi:10.1073/pnas.0610731104.
Ceolotto G, Papparella I, Bortoluzzi A, Strapazzon G, Ragazzo F, Bratti P, et al. Interplay between miR-155, AT1R A1166C polymorphism, and AT1R expression in young untreated hypertensives. Am J Hypertens. 2011;24(2):241–6. doi:10.1038/ajh.2010.211.
Zheng L, Xu CC, Chen WD, Shen WL, Ruan CC, Zhu LM, et al. MicroRNA-155 regulates angiotensin II type 1 receptor expression and phenotypic differentiation in vascular adventitial fibroblasts. Biochem Biophys Res Commun. 2010;400(4):483–8. doi:10.1016/j.bbrc.2010.08.067.
Sethupathy P, Borel C, Gagnebin M, Grant GR, Deutsch S, Elton TS, et al. Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3′ untranslated region: a mechanism for functional single-nucleotide polymorphisms related to phenotypes. Am J Hum Genet. 2007;81(2):405–13. doi:10.1086/519979.
Mottl AK, Shoham DA, North KE. Angiotensin II type 1 receptor polymorphisms and susceptibility to hypertension: a HuGE review. Genet Med: Off J Am Coll Med Genet. 2008;10(8):560–74.
Xu CC, Han WQ, Xiao B, Li NN, Zhu DL, Gao PJ. Differential expression of microRNAs in the aorta of spontaneously hypertensive rats. Sheng Li Xue Bao. 2008;60(4):553–60.
Tome-Carneiro J, Larrosa M, Yanez-Gascon MJ, Davalos A, Gil-Zamorano J, Gonzalvez M, et al. One-year supplementation with a grape extract containing resveratrol modulates inflammatory-related microRNAs and cytokines expression in peripheral blood mononuclear cells of type 2 diabetes and hypertensive patients with coronary artery disease. Pharmacol Res. 2013;72:69–82. doi:10.1016/j.phrs.2013.03.011.
Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58(4):1001–9. doi:10.1002/art.23386.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Author R. Nosalski, Author E. McGinnigle, Author M. Siedlinski and Author T.J. Guzik declare that they have no conflict of interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the authors.
Funding
This study is supported by the National Science Centre of Poland grants: 2011/03/B/NZ4/02454 (T.G.), 2013/11/N/NZ4/00310 (R.N.) and 2012/07/D/NZ4/00644 (M.S.)
Additional information
This article is part of the Topical Collection on Hypertension
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nosalski, R., McGinnigle, E., Siedlinski, M. et al. Novel Immune Mechanisms in Hypertension and Cardiovascular Risk. Curr Cardiovasc Risk Rep 11, 12 (2017). https://doi.org/10.1007/s12170-017-0537-6
Published:
DOI: https://doi.org/10.1007/s12170-017-0537-6