
Abstract
This study develops a simple, cost-effective and sensitive high-performance liquid chromatography with diode array detector (HPLC–DAD) method for the simultaneous determination of eight sulfonylurea herbicides (oxasulfuron, metsulfuron-methyl, triasulfuron, chlorsulfuron, amidosulfuron, mesosulfuron-methyl, bensulfuron-methyl and tritosulfuron) in rapeseed oil. Extraction of target analytes was performed using quick, easy, cheap, effective, rugged and safe-based procedure followed by solid-phase extraction (SPE) and dispersive solid-phase extraction (d-SPE) clean-up, and presents good performance for all of the analytes with recoveries in the range of 67–133% and relative standard deviations (RSD) less than 15%. No significant matrix interference was observed due to the application of effective zirconium dioxide-based sorbent (Z-Sep). Method LOQs for most of the investigated analytes were set at satisfactory low value of 20 ng g−1 in food product. The procedure was evaluated in analyses of actual samples. The most important steps of the method optimization are presented. Novel EMR-Lipid clean-up solution for samples with high fat content was evaluated and compared to Z-Sep sorbent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sulfonylurea herbicides (SUs) belong to the family of acetolactate synthase (ALS) inhibitor herbicides (Brown 1990), and due to their low application rates (10–40 g ha−1), low mammalian toxicity and high herbicidal activity, SUs have become very popular worldwide (Sarmah and Sabadie 2002).
Crop-selective sulfonylurea herbicides have been commercialized for use in wheat, barley, rice, corn, soybeans and oilseed rape (Brown 1990). Ethametsulfuron-methyl is an effective herbicide for the control of wild mustard, stinkweed and other broad-leaved weeds, especially in oilseed rape. However, other SUs, which are used in farming systems to control a range of grass and broadleaf weeds in cereals, can cause considerable damage to non-target crop species such as canola/rapeseed oil. This results in significant limitations in cultivation of rotational crops.
Nevertheless, several commercial seed companies have introduced the imidazolinone-tolerant canola varieties. These Clearfield® canola varieties can grow in rotation where the rotational crop uses imidazolines (ALS inhibitor herbicides) and sulfonylureas and their soil residues do not cause significant injury (Tan et al. 2005). Furthermore, since these varieties were developed using conventional breeding methods, its commercialization is easier than in case of genetically modified (GM) plant, which results in their wide popularity. Another step forward in this matter was the introduction of SU Canola™ (n.d), which is a non-transgenic (non-GMO) sulfonylurea herbicide tolerant canola available in the USA; on track to be available in Canada in 2017; and expected to be launched in other major global markets after 2018. Its introduction provides an alternative for weed control in canola that help manage glyphosate-resistant weeds by using effective SUs. Considering that rapid grow in demand for non-transgenic oil is observed, cultivation of such canola varieties might gain significant popularity and share in total acreage of this crops.
For that reason, reliable analytical methods for determination and quantification of SUs residues in rapeseed oil are indispensable. Pesticide residue analysis in edible oils is still a challenging issue for analysts due to fatty nature of the sample matrix (Masiá et al. 2016). On the one hand, some of the lipids are co-extracted and might cause significant difficulties during subsequent analysis; on the other hand, some fat-soluble non-polar analytes might persist in fatty food sample and give poor extraction efficiency (Rejczak and Tuzimski 2015a). The preparation of oil samples for the determination of pesticides by chromatographic techniques requires the complete removal of the high molecular-mass fat from the sample to maintain the chromatographic system in working order, because ‘dirty’ extracts with even small amounts of fat may harm the columns and detectors (García-Reyes et al. 2007).
To date, extraction of pesticides from edible oil samples is mainly performed by liquid–liquid partitioning with acetonitrile or acetonitrile-hexane mixture (He et al. 2017) and then followed by different clean-up solutions, which seem to be the main limiting step. Gel permeation chromatography (GPC) (Sánchez et al. 2006, Guardia-Rubio et al. 2006), low-temperature precipitation (Li et al. 2007) and different modes of solid phase extraction (SPE) (Husain et al. 2005, López-Feria et al. 2009, Peng et al. 2016) were applied to reduce amount of co-extracted interferences.
Recently, quick, easy, cheap, effective, rugged and safe (QuEChERS) approach has been increasingly applied for pesticide residue analysis in vegetable oils (He et al. 2017). First report on its application to pesticide extraction from olive oil was published in 2007 by Cunha et al. (2007). QuEChERS shows some important advantages such as low solvent consumption, its simplicity and flexibility, as well as high sample throughput. The procedure involves salting-out assisted liquid–liquid partitioning step with acetonitrile followed by extract clean-up by dispersive-solid-phase extraction (d-SPE) (Rejczak and Tuzimski 2015a). Dispersive-SPE allows introduction of different amounts and types of sorbents, so that the procedure can be tailored to cope well with wide scope of the analytes (Rejczak and Tuzimski 2015b). Moreover, a freezing-out step prior to d-SPE has been introduced for initial clean-up of the edible oil extracts (Parrilla Vázquez et al. 2016). Current developments of extraction methods based on modifications to the QuEChERS procedure were elaborated by several authors (Polgár et al. 2012, Ruiz-Medina et al. 2012, Moreno-González et al. 2014, Tuzimski and Rejczak 2016, Parrilla Vázquez et al. 2016, Dias et al. 2016, He et al. 2017).
The identification and quantification of pesticides in fatty samples require suitable selectivity and sensitivity of the analysis. Both gas chromatography (GC) (Guardia-Rubio et al. 2006, López-Feria et al. 2009, Deme et al. 2014, He et al. 2017, Parrilla Vázquez et al. 2016) and liquid chromatography (LC) (Gilbert-López et al. 2010, Sobhanzadeh et al. 2011, Polgár et al. 2012, Moreno-González et al. 2014) coupled with mass spectrometry (MS), or tandem mass spectrometry (MS/MS) detection, are widely used for the determination of pesticides in vegetable oils. Nevertheless, less selective high-performance liquid chromatography with diode array detector (HPLC–DAD) was also successfully applied to the analysis of pesticide residues in edible oils (Jaabiri et al. 2013, Tuzimski and Rejczak 2016, Ma et al. 2016). Stoev and Stoyanov (2007) concluded that the reliability of identification of an analyte by DAD is comparable to the reliability of identification by low resolution MS–MS. It should be emphasized that DAD might be useful in the analysis of samples with complicated matrices when peak purity is determined indicating spectra homogeneity and insignificant influence of co-extracted compounds on proper quantification of the analytes.
The objective of this study was the development and evaluation of simple, cost-effective and robust analytical method based on QuEChERS sample preparation approach followed by HPLC–DAD for the simultaneous analysis of eight SU herbicides in rapeseed/canola oil. Several clean-up methods were evaluated in terms of clean-up efficiency and the satisfactory recovery and precision criteria.
Experimental
Chemicals and Reagents
Pesticide Standards
Standards for the eight sulfonylurea herbicides (SUs) under investigation, such as oxasulfuron, metsulfuron-methyl, triasulfuron, chlorsulfuron, mesosulfuron-methyl, amidosulfuron, bensulfuron-methyl and tritosulfuron were obtained from Pestanal® (Sigma-Aldrich, Supelco, Bellefonte, PA, USA). The standard purity indicated by the manufacturers for all of the reference standards was ≥98%. Individual stock standard solutions (400 mg L−1) were prepared in methanol and were stored at 4 ± 2 °C. A pesticide standard mixture containing all the analytes (10 mg L−1) was prepared by combining suitable aliquots of each individual standard stock solution and diluting them with gradient grade acetonitrile. Physicochemical characteristic and structural formulas of the analytes are presented in Table 1.
Solvents and Mobile-Phase Solutions
Acetonitrile (MeCN) and methanol (MeOH) were pro-chromatography grade and were obtained from E. Merck. Deionized water (0.07–0.09 μS cm−1) was obtained by means of Hydrolab System (Gdansk, Poland) in our laboratory. Formic acid was obtained from POCH (Gliwice, Poland).
QuEChERS Salts and Sorbents and SPE Materials
Anhydrous magnesium sulphate (MgSO4) and sodium chloride (NaCl) were obtained from POCH (Gliwice, Poland). Zirconium dioxide-based sorbents (Z-Sep and Z-Sep Plus) and primary secondary amine (PSA) were purchased from Sigma-Aldrich (Supelco). C18 sorbent (40 μm, Bakerbond) was from J.T. Baker (Deventer, the Netherlands). QuEChERS d-SPE EMR-Lipid (5982–1010) were obtained from Agilent (Folsom, CA, USA). SPE cartridges containing octadecyl sorbent (C18, 2000 mg/6 mL, no. 7020–08) were obtained from Bakerbond (J.T. Baker).
Sample Preparation
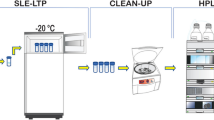
QuEChERS-Based Sample Preparation
Cold-pressed rapeseed oil samples were purchased from local market. For the extraction, 15 g rapeseed oil samples were weighted into 50 mL polypropylene (PP) centrifugation tubes. Then, 10 mL of deionized water was added to the each sample and closed tubes were shaken manually for 30 s. Next, 15 mL of MeCN was added and closed tubes were again vigorously shaken for approximately 1 min. Subsequently, 3 g of NaCl and 6 g of anhydrous MgSO4 were added and the closed tubes were immediately shaken vigorously for approximately 1 min to prevent clumping of the salts. The tubes were centrifuged (Centrifuge MPW-223e, Warsaw, Poland) at 6000 rpm (3480 rcf) for 5 min. The acetonitrile layer (12 mL) of each tube was obtained with a pipette and transferred to the 15 mL (PP) tube.
The proposed sample preparation procedure incorporates two step extract clean-up. Initially, simplified SPE procedure was performed using cartridges containing 2000 mg of C18 sorbent and a Baker SPE-12G SPE chamber (J.T. Baker). Before use, each cartridge was conditioned with 5 mL of MeCN. Then, 12 mL of previously collected acetonitrile extract was loaded into the cartridge and passed through the bed with under pressure about 200–250 mbar. Extracts eluted after SPE were evaporated to dryness in evaporating dishes under a fume hood with air-intake switched on.
The evaporated extracts were reconstituted in 1.2 mL MeCN and transferred into 12-mL PP tubes containing 75 mg Z-Sep (or other sorbents during method optimization). The tubes were shaken vigorously for 1 min and centrifuged as done before. The supernatants (800 μL) were collected and evaporated to dryness under a fume hood. Afterwards, remaining residues were reconstituted in 200 μL of MeCN. Before the analysis via HPLC–DAD, 50 μL of extract was diluted with 50 μL of 50 mM HCOOH (mobile phase component A), which was important to achieve narrower and symmetrical peaks of the analytes. The total analyte enrichment factor for this procedure is equal 20.
Alternative Clean-Up Procedure with EMR-Lipid
Alternatively to classical d-SPE described earlier, rapeseed oil extracts were also cleaned up using EMR-Lipid. For this purpose, the evaporated extracts obtained after initial SPE clean-up were reconstituted in 5 mL of MeCN. Then, 5 mL of water had been added to the EMR-Lipid d-SPE tube prior to addition of the 5 mL of the extract. Subsequently, the mixture was vigorously shaken for 1 min to disperse sample and then centrifuged as done before. Afterwards, a 5-mL aliquot of the supernatant was transferred to a 15 mL EMR-Lipid tube (containing 2 g of salts; 1:4 NaCl/MgSO4) for salting-out step. The contents in the tube were thoroughly shaken for 1 min and centrifuged as done before. Finally, 2 mL of upper acetonitrile layer was collected with a pipette and evaporated to dryness. Afterwards, remaining residues were reconstituted in 200 μL of MeCN. Before the analysis via HPLC–DAD, 50 μL of extract was diluted as done before. The total analyte enrichment factor for the procedure with EMR-Lipid clean-up is equal 12.
RP-HPLC Procedure
Agilent Technologies 1200 HPLC system with a quaternary pump was used for the LC analysis. Analytes were separated using a Scherzo SM-C18 150 mm × 4.6 mm column, with a 3-μm particle size (Imtakt, Portland, OR, USA). The column was thermostated at 22 °C. Mobile phase consisted of 50 mM HCOOH in water (component A) and 50 mM HCOOH in acetonitrile (component B). Gradient elution was applied for the separation of the analytes under investigation with linear gradient of eluent B content starting from 30 to 68.5% in 13 min at 1 mL min−1 flow rate. Final samples were injected onto the column using a Rheodyne manual injector with 20 μL analytical loop. The stationary phase was conditioned between subsequent injections by 10 min (1 mL min−1) with the initial mobile phase composition.
Detection was carried out simultaneously at four different wavelengths (235, 240, 250 and 260 nm). Identification of pesticides was accomplished on the basis of the retention times of the analytes and by comparison between the UV spectra of the reference compounds in the chromatograph library and the UV spectra of the detected peaks in the samples.
Method Validation
HPLC Method Validation
The standard calibration curves of the analytes were constructed by plotting analyte concentration against peak area. Pesticide standards were prepared by combining suitable aliquots of the working standard mixture and diluting them with 50 mM HCOOH in water (mobile phase component A). Standards were injected in triplicates at eight concentrations of 0.2–4 μg mL−1 range under the same chromatographic conditions. The calibration curves of SUs under investigation showed satisfactory linearity and correlation between concentration and peak area over the studied range with the determination coefficients, R 2, ≥0.9998.
The instrumental limits of quantification (LOQ) for all of the analytes were calculated using following formula (1) (International Conference On Harmonisation Of Technical Requirements For Registration Of Pharmaceuticals For Human Use 2005):
where SD is the standard deviation of y-intercept of regression lines (calculated using LINES function in MS Excel 2010), and S is the slope of the calibration plot. Retention times and full calibration data including instrumental LOQs are presented in Table 2.
Recovery and Precision Studies
Rapeseed oil samples were spiked with the herbicides under investigation at three concentrations levels of 20, 50 and 100 ng mL−1. Samples were fortified with the appropriate volume of the working standard mixture and were incubated at room temperature for 1 h before extraction procedure. Recovery studies were performed on the basis of three replicates from the spiking procedure (n = 3) at each concentration level. Relative standard deviations expressed as a percentage (% RSD) were calculated for all of the analytes (Table 3).
Method limits of quantification (mLOQs) were set as the minimum spiking level (ng g−1) that can be quantified with acceptable accuracy and precision (recovery rate in the range of 70 to 120% with RSD% less than 15%) (Table 3).
Matrix Interference Assessment
For the assessment of matrix interference, percentage difference in a signal from the pesticide in matrix compared to the signal in injection solvent was accounted. Matrix interference (MI%) was calculated according to following Eq. (2):
where A denotes peak areas of the standard (A Standard ) and the blank matrix extract (A Post extraction spike ) spiked at the same concentration level (Kruve et al. 2011).
Ideally, value of 0 is related to the absence of matrix interference. Appropriate mixtures of pesticide standards were prepared in 50 mM HCOOH water solution that served for sample dilution (1:1) before analysis via HPLC–DAD. In this way, post-extraction spiked extract was obtained at final concentration of 2, 1 and 0.4 μg mL−1 corresponding to concentration of the analytes in final extract obtained from samples spiked at 100, 50 and 20 ng mL−1, respectively, assuming 100% recovery rate for each analyte. Values of MI% at each fortification levels are presented in Table 3.
Results and Discussion
Separation of the Analytes by HPLC–DAD
Application of Scherzo SM-C18 stationary phase provided sufficient selectivity and chromatographic performance for separation of eight selected SUs, which demonstrate similar structure and physicochemical properties (Table 1). Figure 1 shows chromatogram of the analyte mixture (4 μg mL−1) at 260 nm wavelength. Most important chromatographic parameters were calculated for this analysis automatically by ChemStation® software operating Agilent 1200 series apparatus. Obtained results proved that chromatographic system applied for the analysis demonstrate necessary selectivity and high efficiency, with 236,000 to 669,000 theoretical plates per meter of the column (N/m). All peaks were narrow (width at half height from 0.075 to 0.093 min) and symmetrical, with asymmetry factors (A F) from 1.002 to 1.065 and tailing factors (t F) from 0.946 to 0.995 (Fig. 1). Narrow and symmetrical peaks facilitate proper quantification of the analyte concentration because higher signal to noise (S/N) ratios may be achieved.

Chromatogram (λ = 260 nm) showing separation of eight sulfonylurea herbicides at concentration of 4 μg mL−1 and table showing chromatographic parameters for individual peaks (k capacity factor; Width (min) peak width at half height; A S symmetry factor; t F tailing factor; N/m theoretical plate numbers calculated by half-width method; α selectivity to proceeding peak; R S resolution to proceeding peak calculated by half-width method. Note: All chromatographic parameters were calculated automatically by ChemStation® software operating Agilent 1200 chromatograph; for more details about respective formulas used for calculation, please see the manual linked (https://www.agilent.com/cs/library/usermanuals/Public/G2070-91126_Understanding.pdf). 1 oxasulfuron; 2 metsulfuron-methyl; 3 triasulfuron; 4 chlorsulfuron; 5 amidosulfuron; 6 mesosulfuron-methyl; 7 bensulfuron-methyl; 8 tritosulfuron
Quantitative analysis was based on the calibration curves, which demonstrated high determination coefficients of linear regression (R 2 ≥ 0.9998) for each analyte. Instrumental limits of quantifications (LOQs) were from 33 to 95 ng mL−1, respectively (Table 2). Therefore, HPLC–DAD offers sufficient sensitivity for SUs determination and quantification at trace level, especially when pre-concentration steps are introduced to the sample preparation procedure.
Optimization of QuEChERS-Based Extraction Followed by SPE and d-SPE Clean-Up
Some of the considerations regarding pesticide residue analysis in edible oils earlier published by the authors were taken as a starting point in this study (Tuzimski and Rejczak 2016). Nevertheless, one of the aims was to make the whole procedure relevantly easier and less labour-intensive than the previously reported one.
QuEChERS approach was applied for the extraction of the analytes under investigation. Main alteration to the original method concerned salt addition during partitioning step. It was earlier shown that increased amount of sodium chloride added into the samples helps obtaining higher recoveries of SUs during salting-out liquid–liquid partitioning step of the QuEChERS extraction (Rejczak and Tuzimski 2016). Consequently, in this study, proportion between NaCl and MgSO4 used for this purpose was 1:2 instead of 1:4 applied in the original QuEChERS method. Acetonitrile used as extraction solvent shows limited solubility of lipids; however, significant amounts of non-polar co-extractives are still present in initial QuEChERS extracts of oil samples. Therefore, first clean-up step is based on SPE using C18 sorbent. Acetonitrile extracts were simply passed through the pre-conditioned bed, which allows to get rid of most non-polar interferences. These non-polar compounds were retained on C18 sorbent whereas analytes of interest do not undergo losses during this step.
After this step, obtained extract were colourless in comparison to the acetonitrile layer collected after initial QuEChERS extraction (Fig. 2). This approach reduces labour intensity against full SPE procedure applied in earlier study (Tuzimski and Rejczak 2016). Disposing of the vast majority of non-polar interferences during SPE step facilitates evaporation of extracts, which results in easier implementation of pre-concentration step into the procedure. As a result, tenfold pre-concentration step was at this point applied—12 mL of the extract was evaporated to dryness and residues were then reconstituted in 1.2 mL of MeCN.

Initial QuEChERS extract of rapeseed oil sample (a) and the same extract with 2 g C18 SPE clean-up step (b)
Further extract clean-up was performed on the basis of d-SPE using different types and amounts of sorbents. Proper extract clean-up is especially important in case when simple UV detection using DAD is applied, because limited selectivity of the detector must be supported by adequate interferences removal. Different clean-up solutions were evaluated in terms of interferences removal from the pre-concentrated extracts, recoveries and extraction precision. Taking advantage of previous experience, we assumed that this clean-up step should work well with application of relatively new Z-Sep sorbent. Z-Sep is a sorbent based on modified silica gel with zirconium oxide(IV), which enables Lewis acid/base interactions for efficient interferences removal (Rejczak and Tuzimski 2015b). Up to date, its potential in QuEChERS d-SPE clean-up step was tested in several matrix types, such as different fruits and vegetables (Rajski et al. 2013, Lozano et al. 2014, Tuzimski and Rejczak 2014, Morris and Schriner 2015, Walorczyk et al. 2015, López-Blanco et al. 2016), fish (Sapozhnikova and Lehotay 2013, Kaczyński et al. 2017), edible oils (Moreno-González et al. 2014, Tuzimski and Rejczak 2016, López-Blanco et al. 2016, Dias et al. 2016), Ginko biloba nutraceutical products (Martínez-Domínguez et al. 2015), milk (Rejczak and Tuzimski 2017) and soya milk (Rejczak and Tuzimski 2016), honeybees (Kiljanek et al. 2016) and bee pollen (Parrilla Vázquez et al. 2015).
Three different amounts of 50, 75 and 100 mg of Z-Sep were evaluated (Fig. 3). The best compromise between recovery of the SUs under investigation (at 100 ng g−1 spiking level) and removal of matrix interferences was observed with application of 75 mg of the zirconium-based sorbent. Furthermore, 75 mg of Z-Sep Plus sorbent was also assessed; however, this sorbent strongly retained sulfonylureas whilst no significant improvement in extract clean-up performance was noticed. Addition of small quantities of PSA (15 mg) to Z-Sep with main function to remove more co-extracted constituents such as fatty acids and ionic-lipids was also evaluated. As it was expected, this sorbent retains ionized molecules of weakly acidic SUs by anion exchange mechanism and must be avoided when such analytes are included in the scope of the analysis (Fig. 3). No significant improvement in interferences removal was also shown for 75 mg C18 addition to 75 mg of Z-Sep (Fig. 3).

Graph showing summary of d-SPE clean-up optimization in light of analyte recovery (at 100 ng g−1 fortification level). Error bars indicate RSD% (n = 3)
Additionally, novel EMR-Lipid (enhanced matrix removal—lipid) was taken into evaluation. This clean-up material was so far assessed for analysis of pesticides in kale, pork, salmon and avocado (Han et al. 2016), olive oil, olives and avocado (López-Blanco et al. 2016), as well as edible oils (Dias et al. 2016; Parrilla Vázquez et al. 2016; He et al. 2017). In our study, EMR-Lipid removed less interference than 75 mg of Z-Sep, which is shown in overlaid chromatograms of blank samples obtained after application of both clean-up solutions (Fig. 4). Despite the fact that higher recoveries were obtained with EMR-Lipid than with 75 mg of Z-Sep (Fig. 3), these results cannot be considered optimal due to recovery overestimation caused by the presence of matrix interferences. Dissimilar finding was revealed in study performed by Dias et al. (2016) and Parrilla Vázquez et al. (2016), who found EMR-Lipid superior to Z-Sep sorbent. However, this difference can be explained by the fact that the authors conducted experiments for samples of other edible oils than rapeseed oil, for different analytes, and with application of other instrumentation.

Overlaid chromatograms (λ = 240 nm) of blank rapeseed oil samples after clean up with EMR-Lipid (red) and 75 mg of Z-Sep (blue). Please note that in case of Z-Sep, the extract is \( 1\frac{2\ }{3} \) more concentrated than in case of the procedure with EMR-Lipid, because with EMR-Lipid, only 5 mL aliquot of total supernatant (5 mL of water + 5 mL of extract) after first clean-up step is taken to the second salting-out step
Method Validation
Recovery and Repeatability Study
For recovery studies, three fortification levels were selected: 20, 50 and 100 ng g−1. All recovery experiments were performed in triplicates. Optimized extraction and clean-up procedure combining QuEChERS, SPE on C18 (cartridge containing 2000 mg of the sorbent) and d-SPE with 75 mg of Z-Sep showed good analytical performance in terms of extraction efficiency and repeatability.
Recoveries were in the ranges of 67–103%; 72–119% and 76–133% at 100; and 50 and 20 ng g−1 fortification levels, respectively. In all cases, relative standard deviations expressed as percentage (RSD%; n = 3) were less than 15%, and were in the ranges of 5–12%; 7–13% and 8–14% at 100; and 50 and 20 ng g−1 fortification levels, respectively. Recovery and repeatability data are summarized in Table 3. Exemplary chromatograms obtained from samples fortified at each spiking level are demonstrated in Fig. 5.

Chromatograms (λ = 240 nm) of sulfonylurea herbicide (SUs) standard mixture at concentration of 4 μg mL−1; final extract of un-spiked sample; samples fortified at 100, 50 and 20 ng g−1. 1 oxasulfuron; 2 metsulfuron-methyl; 3 triasulfuron; 4 chlorsulfuron; 5 amidosulfuron; 6 mesosulfuron-methyl; 7 bensulfuron-methyl; 8 tritosulfuron
Recently, Kaczyński (2017) included SUs in the scope of the herbicides investigated in rapeseed oil samples. The author developed QuEChERS-based sample preparation procedure with chitin as a clean-up material followed by LC–MS/MS analysis. Obtained recoveries for SUs were mostly in the range of 80–90%, and were similar to these observed in our study (Kaczyński 2017).
Matrix Interference Assessment
Degree of chemical interference is a pivotal issue in analytical methods that utilize less selective instrumentation. In case of UV detection, the matrix interference takes the form of a direct response overlapping due to the limited selectivity of the detector, because according to the Lambert–Beer’s law, if there are no interactions between the components, the individual absorbances are additive (Rejczak and Tuzimski 2017).
For that reason, in the experiments, authors decided to evaluate MI% using simple approach of standard addition to the blank extracts, which seems to be more reliable in this case. In general, values of 100 ± 20% might be considered as acceptable matrix interference in pesticide residue analysis (document no. SANTE/11945/2015 n.d.). In our study, observed MI% values were from 1 to 12% at 100 ng g−1; 3–18% at 50 ng g−1 and 8–26% at 20 ng g−1 fortification level. Bensulfuron-methyl and tritosulfuron were the analytes that demonstrated the highest MI% values, especially at the lowest spiking level. Obtained results confirm the reliability of the recovery studies of the procedure, but not served for the recovery rates compensation/correction.
What is worth emphasizing is that application of HPLC–DAD allows also continuous quality control of the influence of matrix interferences on proper quantification by obtaining peak purity data of peaks, which refers to a distortion of the analyte spectrum by additional components which partially or completely co-elutes with the major compound of interest (Papadoyannis and Gika 2004, Rejczak and Tuzimski 2016). This is visibly shown in the example of oxasulfuron detected in spiked rapeseed oil samples (Fig. 6). The purity factor of 1000 refers to ideal situation when no interference is observed and spectra within identified peaks demonstrate excellent homogeneity. In sample spiked at 20 ng g−1, the purity factor of oxasulfuron peak was equal 906, which translates to 90.6% purity. In other words, impurity of about 10% was identified, which is in great agreement with MI% of 9% found for this analyte in experimental comparison of signals (peak areas) in solvent-only and final extract samples. Similarly, at 50 and 100 ng g−1, spiking levels purity factors for peaks of oxasulfuron were equal 949 and 985, respectively. These mean impurities of about 5 and 1.5% were identified by peak purity determination, which are in line with MI% of 4 and 2% calculated by standard addition method to the blank extracts (Fig. 6; Table 3).

Diagrams showing matches between UV spectra of oxasulfuron and its library standard, as well as peak purities of its peaks obtained from samples fortified at 20, 50 and 100 ng g−1. Peak purity determination might be useful tool for continuous matrix effect (matrix interference) assessment
Method Limits of Quantification
Document no. SANTE/11945/2015 (n.d.) describes the method limit of quantification (mLOQ) as the minimum concentration which meets the criteria of a mean recovery within the 70–120% range and an RSD ≤ 20%. In our study, oxasulfuron, metsulfuron-methyl, triasulfuron, chlorsulfuron, amidosulfuron and mesosulfuron-methyl meet these criteria at the lowest spiking level of 20 ng g−1. Accordingly, mLOQ values for these sulfonylurea herbicides are set at 20 ng g−1 (Table 3).
For bensulfuron-methyl and tritosulfuron, average recoveries at 20 ng g−1 fortification level exceeded 120% and were equal 133 and 128%, respectively. It may be explained by the presence of matrix interferences co-eluting with this analytes (MI% of 26 and 21%, respectively; Table 3). Therefore, minimum concentration at which these SUs passed validation criteria was 50 ng g−1 and it was selected as their mLOQ values. Nevertheless, detection of bensulfuron-methyl and tritosulfuron is possible at lower concentrations, but one should keep in mind that quantitative results are in this case subjected to errors.
Application to Natural Sample Analysis
Described analytical procedure was evaluated in terms of natural samples analysis. Five different rapeseed oil products were purchased in local markets. Samples were extracted and analysed according to the elaborated QuEChERS/SPE/d-SPE–HPLC–DAD procedure. All tested samples were free from residues of sulfonylurea herbicides under investigation.
Conclusions
In this study, cost-effective and reliable analytical method for analysis of selected sulfonylurea herbicides (SUs) in cold-pressed rapeseed oil samples by HPLC–DAD is proposed. Application of QuEChERS-based extraction followed by SPE and d-SPE clean-up steps enables obtaining satisfactory results in terms of analyte recovery and repeatability (RSD%). Negligible matrix interference was observed due to effective zirconium-based sorbent (Z-Sep) application. Z-Sep showed superior performance that novel enhanced matrix removal of lipids (EMR-Lipid). Peak purity was determined, and we proved that it could be useful tool for matrix interference assessment. Results obtained from peak purity determination are in great agreement with experimental values of degree of chemical interference (MI%) obtained as a percentage difference in signals of analytes in solvent-only and final extract samples.
The procedure has undergone necessary validation study at three spiking levels of 20, 50 and 100 ng g−1. The analytical method was applied for natural sample analysis, and no residues were found. The proposed method fulfils the demand for the analytical procedures for SU investigation in rapeseed oil samples, since novel sulfonylurea tolerant canola/rapeseed varieties appear in the market.
References
Brown HM (1990) Mode of action, crop selectivity, and soil relations of the sulfonylurea herbicides. Pestic Sci 29:263–281. doi:10.1002/ps.2780290304
Cunha SC, Lehotay SJ, Mastovska K, Fernandes JO, Beatriz M, Oliveira PP (2007) Evaluation of the QuEChERS sample preparation approach for the analysis of pesticide residues in olives. J Sep Sci 30:620–632. doi:10.1002/jssc.200600410
Deme P, Azmeera T, Prabhavathi Devi BLA, Jonnalagadda PR, Prasad RBN, Vijaya Sarathi UVR (2014) An improved dispersive solid-phase extraction clean-up method for the gas chromatography–negative chemical ionisation tandem mass spectrometric determination of multiclass pesticide residues in edible oils. Food Chem 142:144–151. doi:10.1016/j.foodchem.2013.07.044
Dias JV, Cutillas V, Lozano A, Pizzutti IR, Fernández-Alba AR (2016) Determination of pesticides in edible oils by liquid chromatography-tandem mass spectrometry employing new generation materials for dispersive solid phase extraction clean-up. J Chromatogr A 1462:8–18. doi:10.1016/j.chroma.2016.07.072
Document N° SANTE/11945/2015 (n.d.): European Commission Directorate-General For Health And Food Safety, Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed, SANTE/11945/2015 http://ec.europa.eu/food/plant/docs/plant_pesticides_mrl_guidelines_wrkdoc_11945_en.pdf (accessed 25.09.2016)
García-Reyes JF, Ferrer C, Gómez-Ramos MJ, Molina-Díaz A, Fernández-Alba AR (2007) Determination of pesticide residues in olive oil and olives. TRAC-Trend Anal Chem 26(3):239–251. doi:10.1016/j.trac.2007.01.004
Gilbert-López B, García-Reyes JF, Fernández-Alba AR, Molina-Díaz A (2010) Evaluation of two sample treatment methodologies for large-scale pesticide residue analysis in olive oil by fast liquid chromatography–electrospray mass spectrometry. J Chromatogr A 1217:3736–3747. doi:10.1016/j.chroma.2010.04.025
Guardia-Rubio M, Fernández-De Córdova ML, Ayora-Caňada MJ, Ruiz-Medina A (2006) Simplified pesticide multiresidue analysis in virgin olive oil by gas chromatography with thermoionic specific, electron-capture and mass spectrometric detection. J Chromatogr A 1108:231–239. doi:10.1016/j.chroma.2006.01.006
Han L, Matarrita J, Sapozhnikova Y, Lehotay SJ (2016) Evaluation of a recent product to remove lipids and other matrix co-extractives in the analysis of pesticide residues and environmental contaminants in foods. J Chromatogr A 1449:17–29. doi:10.1016/j.chroma.2016.04.052
He Z, Wang Y, Wang L, Peng Y, Wang W, Liu X (2017) Determination of 255 pesticides in edible vegetable oils using QuEChERS method and gas chromatography tandem mass spectrometry. Anal Bioanal Chem 409(4):1017–1030. doi:10.1007/s00216-016-0016-9
Husain SW, Kiarostami V, Morrovati M, Tagebakjsj MR, Roozbahani A (2005) Multiresidue determination of organophosphorus pesticide in corn oil using solid-phase extraction with lanthanum silicate sorbent and gas chromatography. J Phys Theor Chem IAU Iran 2(2):57–61
International Conference On Harmonisation Of Technical Requirements For Registration Of Pharmaceuticals For Human Use (2005) Validation of analytical procedures: text and methodology Q2(R1); http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed 23.10.2016)
Jaabiri I, Belhaj D, Turki N, Kallel M, Ayadi H, Ksantini M, Bouzid J, Gargouri R (2013) Development and method validation for determination of Deltamethrin residue in olive oil using a reversed-phase high performance liquid chromatography. IOSR-JAC 6(3):1–8
Kaczyński P (2017) Large-scale multi-class herbicides analysis in oilseeds by rapid one-step QuEChERS-based extraction and cleanup method using liquid chromatography–tandem mass spectrometry. Food Chem 230:411–422. doi:10.1016/j.foodchem.2017.03.076
Kaczyński P, Łozowicka B, Perkowski M, Szabuńko J (2017) Multiclass pesticide residue analysis in fish muscle and liver on one-step extraction-cleanup strategy coupled with liquid chromatography tandem mass spectrometry. Ecotoxicol Environ Saf 138:179–189. doi:10.1016/j.ecoenv.2016.12.040
Kiljanek T, Niewiadowska A, Semeniuk A, Gaweł M, Borzęcka M, Posyniak M (2016) Multi-residue method for the determination of pesticides and pesticide metabolites in honeybees by liquid and gas chromatography coupled with tandem mass spectrometry—honeybee poisoning incidents. J Chromatogr A 1435:100–114. doi:10.1016/j.chroma.2016.01.045
Kruve A, Herodes K, Leito I (2011) Accounting for matrix effects of pesticide residue liquid chromatography/electrospray ionisation mass spectrometric determination by treatment of background mass spectra with chemometric tools. Rapid Commun Mass Spectrom 25:1159–1168. doi:10.1002/rcm.4971
Li L, Zhou Z, Pan C, Qian C, Jiang S, Liu F (2007) Determination of organophosphorus pesticides in soybean oil, peanut oil and sesame oil by low-temperature extraction and GC-FPD. Chromatographia 66(7/8):625–629. doi:10.1365/s10337-007-0349-9
López-Blanco R, Nortes-Méndez R, Robles-Molina J, Moreno-González D, Gilbert-López B, García-Reyes JF, Molina-Díaz A (2016) Evaluation of different cleanup sorbents for multiresidue pesticide analysis in fatty vegetable matrices by liquid chromatography tandem mass spectrometry. J Chromatogr A 1456:89–104. doi:10.1016/j.chroma.2016.06.019
López-Feria S, Cárdenas S, Valcárcel M (2009) One step carbon nanotubes-based solid-phase extraction for the gas chromatographic–mass spectrometric multiclass pesticide control in virgin olive oils. J Chromatogr A 1216:7346–7350. doi:10.1016/j.chroma.2009.02.060
Lozano A, Rajski Ł, Uclés S, Belmonte-Valles N, Mezcua M, Fernández-Alba AR (2014) Evaluation of zirconium dioxide-based sorbents to decrease the matrix effect in avocado and almond multiresidue pesticide analysis followed by gas chromatography tandem mass spectrometry. Talanta 118:68–83. doi:10.1016/j.talanta.2013.09.053
Ma F, Wu R, Li P, Yu L (2016) Analytical approaches for measuring pesticides, mycotoxins and heavy metals in vegetable oils: a review. Eur J Lipid Sci Technol 118:339–352. doi:10.1002/ejlt.201400535
Martínez-Domínguez G, Romero-González R, Garrido Frenich A (2015) Determination of toxic substances, pesticides and mycotoxins, in Ginkgo biloba nutraceutical products by liquid chromatography Orbitrap-mass spectrometry. Microchem J 118:124–130. doi:10.1016/j.microc.2014.09.002
Masiá A, Morales Suarez-Varela M, Llopis-Gonzalez A, Picó Y (2016) Determination of pesticides and veterinary drug residues in food by liquid chromatography-mass spectrometry: a review. Anal Chim Acta 936:40–61. doi:10.1016/j.aca.2016.07.023
Moreno-González D, Huertas-Pérez JF, García-Campaña AM, Gámiz-Gracia L (2014) Determination of carbamates in edible vegetable oils by ultra-high performance liquid chromatography-tandem mass spectrometry using a new clean-up based on zirconia for QuEChERS methodology. Talanta 128:299–304. doi:10.1016/j.talanta.2014.04.045
Morris BD, Schriner RB (2015) Development of an automated column solid-phase extraction cleanup of QuEChERS extracts, using a zirconia-based sorbent, for pesticide residue analyses by LC-MS/MS. J Agric Food Chem 63:5107–5119. doi:10.1021/jf505539e
Papadoyannis IN, Gika HG (2004) Peak purity determination with a diode array detector. J Liq Chromatogr R T 27(6):1083–1092. doi:10.1081/JLC-120030180
Parrilla Vázquez P, Lozano A, Uclés S, Gómez Ramos MM, Fernández-Alba AR (2015) A sensitive and efficient method for routine pesticide multiresidue analysis in bee pollen samples using gas and liquid chromatography coupled to tandem mass spectrometry. J Chromatogr A 1426:161–173. doi:10.1016/j.chroma.2015.11.081
Parrilla Vázquez P, Hakme E, Uclés S, Cutillas V, Martínez Galera M, Mughari AR, Fernández-Alba AR (2016) Large multiresidue analysis of pesticides in edible vegetable oils by using efficient solid-phase extraction sorbents based on quick, easy, cheap, effective, rugged and safe methodology followed by gas chromatography–tandem mass spectrometry. J Chromatogr A 1463:20–31. doi:10.1016/j.chroma.2016.08.008
Peng X-T, Li Y-N, Xia H, Peng L-J, Feng Y-Q (2016) Rapid and sensitive detection of fipronil and its metabolites in edible oils by solid-phase extraction based on humic acid bonded silica combined with gas chromatography with electron capture detection. J Sep Sci 39:2196–2203. doi:10.1002/jssc.201501250
Pesticide Properties Database (PPDB) (n.d.) http://sitem.herts.ac.uk/aeru/ppdb/en/. Accessed 17 Jan 2017
Polgár L, Kmellár B, García-Reyes JF, Fodor P (2012) Comprehensive evaluation of the clean-up step in QuEChERS procedure for the multi-residue determination of pesticides in different vegetable oils using LC-MS/MS. Anal Methods 4:1142–1148. doi:10.1039/c2ay05543k
Rajski Ł, Lozano A, Uclés A, Ferrer C, Fernández-Alba AR (2013) Determination of pesticide residues in high oil vegetal commodities by using various multi-residue methods and clean-ups followed by liquid chromatography tandem mass spectrometry. J Chromatogr A 1304:109–120. doi:10.1016/j.chroma.2013.06.070
Rejczak T, Tuzimski T (2015a) A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem 13:980–1010. doi:10.1515/chem-2015-0109
Rejczak T, Tuzimski T (2015b) Recent trends in sample preparation and liquid chromatography/mass spectrometry for pesticide residue analysis in food and related matrixes. J AOAC Int 98(5):1143–1162. doi:10.5740/jaoacint.SGE1_Rejczak
Rejczak T, Tuzimski T (2016) Simple, cost-effective and sensitive liquid chromatography diode array detector method for simultaneous determination of eight sulfonylurea herbicides in soya milk samples. J Chromatogr A 1473:56–65. doi:10.1016/j.chroma.2016.10.023
Rejczak T, Tuzimski T (2017) QuEChERS-based extraction with dispersive solid phase extraction clean-up using PSA and ZrO2-based sorbents for determination of pesticides in bovine milk samples by HPLC-DAD. Food Chem 217:225–233. doi:10.1016/j.foodchem.2016.08.095
Ruiz-Medina A, Llorent-Martínez EJ, Fernández-de Córdova ML, Ortega-Barrales P (2012) Automated optosensor for the determination of carbaryl residues in vegetable edible oils and table olive extracts. J Food Compos Anal 26:66–71. doi:10.1016/j.jfca.2012.02.003
Sánchez AG, Ramos Martos N, Ballesteros E (2006) Multiresidue analysis of pesticides in olive oil by gel permeation chromatography followed by gas chromatography–tandem mass-spectrometric determination. Anal Chim Acta 558:53–61. doi:10.1016/j.aca.2005.11.019
Sapozhnikova Y, Lehotay SJ (2013) Multi-class, multi-residue analysis of pesticides, polychlorinated biphenyls, polycyclic aromatic hydrocarbons, polybrominated diphenyl ethers and novel flame retardants in fish using fast, low-pressure gas chromatography–tandem mass spectrometry. Anal Chim Acta 758:80–92. doi:10.1016/j.aca.2012.10.034
Sarmah AK, Sabadie J (2002) Hydrolysis of sulfonylurea herbicides in soils and aqueous solutions: a review. J Agric Food Chem 50:6253–6265. doi:10.1021/jf025575p
Sobhanzadeh E, Bakar NKA, Abas MRB, Nemati K (2011) An efficient extraction and clean-up procedure for pesticide determination in olive oil. Eur J Lipid Sci Technol 113:862–869. doi:10.1002/ejlt.201000384
Stoev G, Stoyanov A (2007) Comparison of the reliability of the identification with diode array detector and mass spectrometry. J Chromatogr A 1150:302–311. doi:10.1016/j.chroma.2006.12.026
SU Canola™ (n.d.) (Cibus, San Diego, CA, USA) https://www.cibus.com/products.php (accessed on-line 10.12.2016)
Tan S, Evans RR, Dahmer ML, Singh BK, Shaner DL (2005) Imidazolinone-tolerant crops: history, current status and future. Pest Manag Sci 61:246–257. doi:10.1002/ps.993
Tuzimski T, Rejczak T (2014) Determination of pesticides in sunflower seeds by high-performance liquid chromatography coupled with a diode array detector. J AOAC Int 97(4):1012–1020. doi:10.5740/jaoacint.SGETuzimski
Tuzimski T, Rejczak T (2016) Application of HPLC–DAD after SPE/QuEChERS with ZrO2-based sorbent in d-SPE clean-up step for pesticide analysis in edible oils. Food Chem 190:71–79. doi:10.1016/j.foodchem.2015.05.072
Walorczyk S, Drożdżyński D, Kierzek R (2015) Two-step dispersive-solid phase extraction strategy for pesticide multiresidue analysis in a chlorophyll-containing matrix by gas chromatography–tandem mass spectrometry. J Chromatogr A 1412:22–32. doi:10.1016/j.chroma.2015.08.022
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Tomasz Tuzimski declares that he has no conflict of interest. Tomasz Rejczak declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rejczak, T., Tuzimski, T. Method Development for Sulfonylurea Herbicides Analysis in Rapeseed Oil Samples by HPLC–DAD: Comparison of Zirconium-Based Sorbents and EMR-Lipid for Clean-up of QuEChERS Extract. Food Anal. Methods 10, 3666–3679 (2017). https://doi.org/10.1007/s12161-017-0939-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-017-0939-6