
Abstract
Lignin is a key factor limiting saccharification of lignocellulosic feedstocks. In this comparative study, various lignin methods—including acetyl bromide lignin (ABL), acid detergent lignin (ADL), Klason lignin (KL), and modified ADL and KL determination methods—were evaluated for their potential to assess saccharification efficiency. Six diverse accessions of the bioenergy crop miscanthus were used for this analysis, which included accessions of Miscanthus sinensis, Miscanthus sacchariflorus, and hybrid species. Accessions showed large variation in lignin content. Lignin estimates were different between methods, but (highly) correlated to each other (0.54 ≤ r ≤ 0.94). The strength of negative correlations to saccharification efficiency following either alkaline or dilute acid pretreatment differed between lignin estimates. The strongest and most consistent correlations (−0.48 ≤ r ≤ −0.85) were obtained with a modified Klason lignin method. This method is suitable for high throughput analysis and was the most effective in detecting differences in lignin content (p < 0.001) between accessions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Biomass is an abundant source of carbon that can be used for the production of biofuels. This carbon is an important basic element of the different plant components including the cell walls, which are mainly composed of the structural polysaccharide cellulose, hemicellulosic polysaccharides, and the aromatic polymer lignin [1–4]. The conversion of biomass into biofuel depends on the enzymatic saccharification of structural polysaccharides into their monosaccharide building blocks, which can be subsequently fermented into bioethanol.
Lignin is one of the key components limiting the conversion of biomass into biofuel. It cross-links to hemicellulosic polysaccharides to form a highly impermeable matrix that imparts strength to the plant cell wall and shields cellulose—the main source of fermentable sugars—from chemical and enzymatic hydrolysis [5–8]. In addition, it impedes the efficiency of enzymatic saccharification by irreversibly adsorbing hydrolytic enzymes, which renders them ineffective [9, 7]. As lignin is one of the most important barriers in the conversion of biomass into biofuels, reducing lignin content (or altering its composition) in bioenergy crops is critical to reduce processing costs and increase the cost-competitiveness of cellulosic biofuels [10, 11].
Lignin is chemically described as a heteropolymer of phenylpropanoids, primarily p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol, although a variety of other compounds are incorporated in smaller quantities [6, 12, 13]. Polymerization of these subunits occurs in the cell wall via oxidative radicalization reactions that lead to a large number of different coupling products and bonds, some of which are difficult to break [13–15]. Similar radical coupling reactions incorporate ferulate monomers and dimers into lignin, which, via diferulate cross-linking, give rise to cross-links between lignin and feruloylated hemicellulose side-chains that anchor lignin onto the cell wall polysaccharides [4]. These chemical characteristics and its extensive cross-linking to hemicellulose render lignin an intractable component. Lignin is hydrophobic due to its aromatic nature and has a high tendency to self-associate [15, 16]. As a result, the isolation, structural characterization, and quantification of the complete native lignin polymer are challenging [15].
Nonetheless, a number of different methods for determining lignin content in biomass samples have been developed over the past decades, primarily for their application in paper making and animal nutrition. These methods can be grouped into two distinct categories, gravimetric methods, and spectrophotometric methods [14, 16, 17]. Gravimetric methods are the most widely used in both research and industry. The most common and one of the oldest methods to quantify lignin content is the Klason method, developed in the early 1900s by Klason. It is based on the hydrolysis of cell wall polysaccharides using sulphuric acid and gravimetric determination of the acid insoluble residue as Klason lignin (KL) [17, 18]. To deal with concerns related to the contamination of KL with protein, Van Soest proposed to include an acid detergent extraction step to remove proteins (and some other components) prior to polysaccharide hydrolysis, which was particularly useful for forage samples containing high protein concentrations [19–21]. This lignin quantification method—measuring acid detergent lignin (ADL)—is widely used for evaluating feed quality in forage grasses. It is adopted in a sequential fiber analysis protocol that is based on the sequential solubilization of cell wall fractions using neutral detergent, acid detergent, and sulfuric acid solutions [19–21]. While this method is widely employed for evaluating the nutritive value of forages, it may underestimate lignin concentration, as a fraction of the lignin is solubilized during the detergent and acid digestion reactions [22, 23].
Spectrophotometric methods are based on the solubilization of lignin from the cell wall and subsequent determination of its specific absorbance at certain wavelengths. To enable the solubilization of lignin, this polymer must be derivatized, which is most commonly accomplished using acetyl bromide, leading to the determination of acetyl bromide lignin (ABL) [16, 18, 24]. These spectrophotometric lignin determination lignin methods may suffer from interference of light absorption by other biomass components at the same wavelengths, leading to an overestimation of lignin content [16]. Furthermore, these methods require a well-defined lignin standard to calibrate the estimation of lignin concentration from optical density measurements [16, 25].
None of the currently available lignin methods is considered a standard unambiguous method for determining lignin content, as concerns exist for each of these lignin methods related to their under or overestimation of lignin content [15, 16]. More problematically, while all these methods are widely used, large discrepancies are reported in the different estimates of lignin content between them [23, 26–32]. In a recent study, Fukushima and Hatfield compared the performance of some lignin methods on a number of different plant samples and reported on average a twofold difference in lignin content between the ABL method and the ADL method [29]. In specific plant samples, however, even more than a fourfold difference between the two methods was reported, emphasizing that differences between lignin concentrations obtained using different methods are not systematic and directly convertible, but rather dependent on the sample being analyzed. If the ratio between different lignin measurements is variable between samples, this suggests that different lignin methods are measuring different fractions of the native lignin polymer and that these fractions are variable between samples.
Recently, Moreira-Vilar and co-workers compared a number of different lignin methods and concluded that the ABL method outperformed the other methods, primarily because it gave the highest estimates of lignin content [26]. However, considering that lignin is usually quantified because of its effect on the efficiency of a certain process, for example, paper production, ruminant digestion, and saccharification for biofuel production, selecting the most appropriate lignin method may depend on which estimate of lignin content has the highest predictive ability of biomass quality for a certain application. Some recent studies, for example, evaluated different lignin methods in forage samples for their predictive ability of digestibility [27, 29, 32]. Fukushima and Hatfield concluded from their comparative study that ABL provided stronger and more consistent negative correlations to digestibility characters than the other methods [29].
Discrepancies in the utilization of lignin content (measured through different methodologies) as an indicator of biomass quality could result from the fact that lignin extraction procedures act differently on lignin types with different monomeric composition or that possess different types and numbers of crosslinks between lignin and other cell wall components. Although such factors might underpin differences in recalcitrance between different lignin fractions, first, it must be established if different lignin fractions have different implications for saccharification of miscanthus biomass.
Despite the implications of lignin as a major recalcitrance factor in bioenergy conversion technologies, to our knowledge no studies have been performed to compare the different lignin methods for their predictive ability of saccharification efficiency in potential bioenergy feedstocks. Consequently, it is possible that the limited resources available for compositional analysis of biomass feedstocks for fuel production are spent on lignin analysis using a method that is not optimal for this evaluation. Similarly, selection of genotypes with reduced lignin content in a bioenergy crop breeding program might be more effective in improving saccharification efficiency using a certain lignin determination method.
In the manuscript, various lignin methods, including ABL, KL, and ADL, are compared for their applicability to assess the potential of bioenergy feedstocks. To this end, their predictive ability of saccharification efficiency and their ability to discriminate between genotypes are evaluated in a diverse set of accessions of the important bioenergy crop miscanthus.
Materials and Methods
Plant Materials
Six different miscanthus accessions, belonging to three different miscanthus species, were used in this study; two accessions of Miscanthus sinensis, including the commercial cultivar ‘Goliath’, two of Miscanthus sacchariflorus, including the commercial cultivar ‘Robustus’, and two clones derived from crosses between the two species, including the commercially-used clone known as Miscanthus x giganteus (Supplementary Table S1). The accessions were grown in Wageningen, the Netherlands, in a field trial with a randomized block design with three replications. The field trial was established in May 2012. The planting material used to establish the trial was produced clonally by in vitro propagation, except for one accession (OPM-13), which consisted of seed-derived plants resulting from a cross between two M. sinensis parents. A total of 49 plantlets were planted per plot with a density of two plants m, resulting in a plot size of 25 m2. To avoid influence of a potential border effect, only the inner nine plants within plots were harvested for analysis in March 2013. After harvesting, the plant shoots were stripped from leaves and the remaining stems were chopped and air dried at 60 °C for 72 h. The dried stem material was ground using a hammer mill with a 1-mm screen.
Compositional Analysis
Ground stem samples from the field trial were used for compositional analysis. An overview of compositional characteristics determined in this study is provided in Supplementary Table S2. All samples were measured in quadrupole.
Cell Wall Carbohydrate Content
Neutral detergent fiber (NDF) and acid detergent fiber (ADF) contents of stem dry matter were determined according to protocols developed by Ankom Technology (ANKOM Technology Corporation, Fairpoint, NY), which are essentially based on the work of Goering and Van Soest [19–21]. Detergent fiber contents were subsequently used to obtain estimates for the contents of cell wall (CW), cellulose (Cel), and hemicellulosic polysaccharides (Hem) in stem dry matter, as described in Supplementary Table S2.
Acid Detergent Lignin
The residue of each ADF analysis was used for the determination of ADL, according to the protocol developed by Ankom Technology (ANKOM Technology Corporation, Fairpoint, NY), which is based on the work of Goering and Van Soest [19–21, 33]. It comprised a 3-h hydrolysis in 72 % H2SO4 in 1 L laboratory bottles that were placed horizontally on an orbital shaker set at 160 rpm. After hydrolysis, the samples were extensively washed with deionized water and dried for 12 h at 103 °C. The remaining sample residue is considered as acid detergent lignin and was gravimetrically determined. In parallel, an alternative method was also tested, in which the ADL determination was performed on sample material that was sequentially subjected to both a neutral and an acid detergent extraction treatment. Sequential determinations of NDF, ADF, and ADL fastens the procedure and results in a purer ADL fraction, from which xylans and pectins have been removed [19, 16]. The lignin content determined in this way is henceforth referred to in this study as ADLseq.
Acetyl Bromide Lignin
The residual material from each of the four NDF determinations per sample were pooled and used as basis for determining ABL following the method described by Fukushima and Kerley [34]. A single ABL determination comprised precise weighing of 20–25 mg of NDF material into a 2-ml Eppendorf tube. The sample was then digested using 1.5 ml 25 % (v/v) acetyl bromide in acetic acid for 2 h at 50 °C and constant shaking (800 rpm). After digestion of the sample, tubes were centrifuged for 5 min at 13,000 rpm. A 15 μl aliquot of the solution was then added to an Eppendorf tube containing 200 μl of 0.3 M NaOH and 685 μl of acetic acid. Finally, 100 μl 0.5 M hydroxylamine hydrochloride was added and after exactly 30 min the optical density of the solution was measured in duplicate at 280 nm against a blank containing all chemical reagents, but no sample material. Acetyl bromide lignin concentration (mg/ml) was then calculated using a regression Eq. (1):
where A is the average optical density reading of the two measurements; 0.0009 is the mean intercept value of the regression equation as determined by Fukushima and Kerley [34]; DF is the dilution factor, 100×; 17.78 is the miscanthus-specific extinction coefficient as determined by Lygin et al. [35]; and Sample is the amount of NDF material in mg.
Klason Lignin
The same pooled NDF residues used as starting material for the ABL analyses were used for the determination of KL. This was done according to the Laboratory Analytical Procedure “Determination of Structural Carbohydrates and Lignin in Biomass”, a two-step acid hydrolysis method developed by the National Renewable Energy Laboratory (NREL) [36]. The procedure started with the hydrolysis of 300 mg of NDF material in 3 ml 72 % H2SO4 in a 100-ml glass pressure tube (Ace Glass Inc., Vineland, NJ) for 1 h at 30 °C with constant shaking (160 rpm). After 1 h, the acid concentration was diluted to 4 % by adding 84 ml deionized water, after which samples were hydrolyzed by autoclaving the tubes at 121 °C for 1 h. After cooling down, the samples were vacuum-filtered using glass filtering crucibles (30 ml, P4, Klaus Hofman, Staudt, Germany). The residue was dried for 12 h at 103 °C and weighed for the determination of Klason lignin. A separate experiment was conducted to test the feasibility of analyzing smaller biomass samples, with the aim of increasing the throughput of the KL method. In this modified KL method, the sample and reagent quantities were reduced 10-fold, while all other process steps remained the same. The Klason lignin results of this down-scaled experiment are reported as ds-KL.
Acid Soluble Lignin
The filtrate obtained from the vacuum-filtration step of the Klason lignin determination was captured and purified using 0.45 μm filters to quantify acid soluble lignin (ASL). ASL was determined spectrophotometrically at 205 nm using quartz cuvettes. ASL concentrations were calculated using Eq. (2) [37]:
where A is the absorption value; V is the hydrolyzate volume; DF is the dilution factor, 20×; K is the absorptivity constant, 110 L/g/cm as determined by Xu et al. [38]; and Sample is the amount of NDF material in mg.
Neutral Sugar Contents
The filtrate obtained from the vacuum-filtration step of the Klason lignin determination was captured and purified using 0.45 μm filters to quantify the amount of neutral sugars released from cell wall samples. Two different dilutions were made, one for determining the content of glucose and xylose (dilution factor 50) and another for determining the arabinose content (dilution factor 10). Neutral sugar contents were determined by high performance anion exchange chromatography (HPAEC) analysis on a Dionex system equipped with a CarboPac PA1 column and a pulsed amperometric detector (Dionex, Sunnydale, CA). The ratio of arabinose to xylose was also determined, which constitutes an estimate of the degree of hemicellulose substitution (DHS) [39].
Saccharification Efficiency
Separate analyses of ground stem samples were performed for the characterization of saccharification efficiency. Saccharification reactions were carried out using three 500 mg subsamples per stem sample. All samples were briefly treated with α-amylase and repeatedly washed with deionized water (3×, 5 min, ~60 °C) in order to remove all interfering stem soluble sugars. The remaining biomass was then subjected to either an alkaline pretreatment or a dilute acid (DA) pretreatment. Alkaline pretreatments were carried out in 50 ml plastic centrifuge tubes with 15 ml 2 % NaOH at 50 °C with constant shaking (160 rpm) for 2 h in an incubator shaker (Innova 42, New Brunswick Scientific, Enfield, CT). Dilute acid pretreatments were carried out in custom-built stainless steel reactors, essentially as described by Torres et al. [40]. Briefly, pretreatment comprised hydrolysis in 15 ml of 0.17 % (w/v) H2SO4 at 140 °C for 30 min in a temperature controlled oil bath. After 30 min, the reactions were quickly quenched by submerging the reactors in a cold water bath.
The conditions chosen for pretreatment were fairly mild. In this study, the objective of the pretreatment was not to maximize sugar yields but to use conditions that better discriminate genotypic differences in the release of sugars following the combined operations of pretreatment and enzymatic saccharification. The severity (log M0) of the pretreatment was 1.78 for the sodium hydroxide pretreatment and 1.99 for the dilute sulphuric acid pretreatment, as calculated by the following Eq. (3) [41]:
where t is the reaction time; C is the concentration of chemical catalyst (%w/v); n is the empirically determined constant fitted to be 0.849 and 3.90 for sodium hydroxide and sulfuric acid, respectively, [41, 42]; and T is the reaction temperature in °C.
Pretreated samples were then washed to neutral pH with deionized water (2×, 5 min, 50 °C) and with 0.1 M sodium citrate buffer (pH 4.6, 5 min, 50 °C). Saccharification reactions were subsequently carried out according to the NREL Laboratory Analytical Procedure “Enzymatic saccharification of lignocellulosic biomass” [43]. Pretreated samples were hydrolyzed for 48 h with 300 μl of the commercial enzyme cocktail Accellerase 1500 (DuPont Industrial Biosciences, Leiden, NL) supplemented with 15 μl endo-1,4-β-xylanase M1 (Megazyme, Bray, IE) in an incubator shaker (Innova 42, New Brunswick Scientific, Enfield, CT) set at 50 °C and constant shaking (160 rpm). These enzymes combined have the following specific activities: endoglucanase 2200–2800 CMC U/g, beta-glucosidase 450–775 pNPG U/g, and endoxylanase 230 U/mg. Reactions were carried out in 44 ml 0.1 M sodium citrate buffer (pH 4.6), containing 0.4 ml 2 % sodium azide to prevent microbial contamination.
The release of sugars during dilute acid pretreatment and enzymatic saccharification reactions was analyzed by HPAEC as described previously for neutral sugars. Saccharification efficiency was assessed by the respective percentages of glucose and xylose released from the biomass samples by the combined actions of pretreatment and enzymatic saccharification (Supplementary Table S2).
Statistical Analysis
General analyses of variance (ANOVA) were performed for all traits taking into account the randomized block design of the field trial using the following Eq. (4):
where Y ij is the response variable, μ is the overall mean, β 1 X i is the contribution of the genotype, and β 2 X j is the block effect.
Multiple comparisons analysis was performed to distinguish significant (p < 0.05) genotypic differences using Fisher’s protected least significant difference (LSD) test on genotype means. Correlation analysis was performed to identify the significance (p < 0.05), strength and direction of interrelationship between sample characteristics using Pearson’s correlation coefficients. All statistical analyses were performed using Genstat for Windows, 14th edition software package (VSN International, Hemel Hempstead, UK).
Results and Discussion
Characterization of Cell Wall Carbohydrate Content and Composition of Six Diverse Miscanthus Accessions
Ground miscanthus stem samples were analyzed for cell wall compositional characteristics. The results are presented in Table 1 as accession means for neutral and acid detergent fiber, structural carbohydrate, and neutral sugar contents. As expected for miscanthus harvested at complete senescence, low moisture and high cell wall contents were observed [44–46]. On average, approximately 85 % of the dry biomass consisted of cell wall material, ~46 % consisted of cellulose and ~31 % consisted of hemicellulosic polysaccharides. Within the panel of miscanthus accessions, large variation was present in the contents of cellulose, ranging from ~43 to ~48.5 %, and hemicellulosic polysaccharides, ranging from ~27 to ~34 %. Glucose and xylose were the most abundant monosaccharides and accounted for almost 70 % of the cell wall material. A minor fraction of the monosaccharides consisted of arabinose, which made up on average less than 2 % of the cell wall material. Between the species of miscanthus, the M. sacchariflorus accessions showed the highest contents of cellulose, glucose, and xylose, whereas the M. sinensis accessions had the highest contents of hemicellulosic polysaccharides and arabinose. Hybrid accessions were intermediate for cellulose content, but had the lowest fraction of hemicellulosic polysaccharides. OPM-4 was the accession that had the highest amount of glucose potentially available for saccharification reactions, with both the highest cell wall content (87.06 %) and the highest cell wall glucose content (43.96 %).
Large Differences Observed Between the Various Lignin Methods
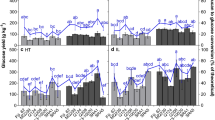
The lignin contents of miscanthus stem samples were evaluated using different lignin methods. The comparative study showed large differences in lignin content between the three most commonly used methods, ABL, KL, and ADL (Fig. 1). The highest lignin contents were obtained by the ABL method, ranging from ~16 to 22 %. Values obtained by the KL method were slightly lower and ranged from ~13 to 20 %. Considerably, lower estimates were obtained by the ADL method, which ranged from ~7 to 14 %. Such striking differences were anticipated and are consistent with the results of previously published comparisons of these lignin methods [23, 26, 27, 29, 32, 47]. The most likely explanation for the low lignin concentrations obtained by the ADL method is the loss of lignin during the acid detergent extraction of the method [23, 32].

Lignin content in miscanthus stem samples as determined by the acetyl bromide lignin (ABL), the acid detergent lignin (ADL), and the Klason lignin (KL) methods, as well as modified versions of the ADL and the KL method. Plotted lines are regression lines of the plot lignin contents on the average plot lignin content as determined by ADL, ABL, and KL
The mean values for each accession for the different lignin methods are reported in Table 2. The overall means over accessions were 18.66 % for ABL, 16.61 % for KL, and 9.87 % for ADL. Despite such differences in lignin estimates between the methods, all lignin methods, except the ASL method, consistently identified OPM-9 (M. x giganteus) as the accession with the highest lignin content and OPM-2 as the accession with the lowest lignin content. Lignin concentrations also displayed a similar trend between species, with the hybrid accessions generally having higher lignin contents than M. sinensis and with M. sacchariflorus accessions generally having the lowest lignin contents (Table 2).
In addition to the three most commonly used lignin measurements, three additional lignin measurements were obtained on the same samples: a modified (sequential) ADL, a modified (down-scaled) Klason lignin and acid soluble lignin (ASL) measurement. Both modified versions of the ADL and the KL protocol resulted in lower lignin estimates compared to their respective reference methods (Fig. 1, Table 2). The sequential variant of the ADL method resulted in an overall mean lignin content over accessions of 6.30 %, which was ~3.5 % lower than that of the conventional ADL method. The additional neutral detergent extraction step in the modified ADL method thus resulted in a loss of lignin compared to the reference protocol. For the modification of the Klason method, an overall mean of 14.48 % was found, which was ~2.1 % lower than the reference (Table 2). The reason why the down-scaled protocol resulted in a lower estimate of lignin remains unclear, as biomass samples in both protocols receive the same treatment. However, similar observations were made by Ibáñez et al. (2014) upon down-scaling the Klason method, so it is unlikely to be due to a technical error [48]. The third additional measurement was the quantification of ASL in the hydrolyzate resulting from the sulfuric acid hydrolysis reactions in the Klason method. For all samples, the acid soluble fraction of the lignin was small compared to the amount of acid insoluble lignin. Accession means ranged from 3.23 to 3.85 %, with an overall mean over accessions of 3.69 %.
Large variation was present among the accessions in lignin estimates derived from the different lignin determination methods, and all methods were able to uncover statistically significant differences among them (Tables 2 and 3). The most significant differences (p < 0.001) among accessions were found using the ADL method and the modified KL method (Table 3). Both methods also resulted in a large range in the performance of accessions (4.71 % for ADL and 4.56 % for ds_KL). The other methods resulted in smaller ranges of variation in lignin estimates (<4.07) and lower discriminative abilities among accessions (0.002 ≤ p ≤ 0.018), making them less suitable for screening small differences between accessions.
Correlation analysis was used to investigate the interrelationships between the different lignin estimates (Fig. 2). This analysis showed that despite the large differences in the estimation of lignin between different lignin determination methods, the estimates of lignin content were correlated (0.54 ≤ r ≤ 0.94). Most notably, ADL and KL were more strongly correlated to each other (r = 0.87) than they were to ABL (r = 0.63 and 0.54, respectively). A reason for this might be the similarities in the way these lignin estimates are determined. ADL and KL concentrations are both determined by weighing the remaining residue after acid-catalyzed hydrolysis of the cell wall polysaccharides, whereas ABL concentration is determined through spectrophotometric quantification of solubilized lignin.

Correlations among lignin estimates obtained using different lignin determination methods. Only Pearson correlation coefficients that differed significantly from zero (p > 0.05) are reported
In addition, the correlation analysis showed that the lignin estimates obtained by the modified Klason and modified acid detergent protocols tested in this study were highly correlated (r = 0.87 and 0.91, respectively) to lignin estimates obtained by their corresponding reference methods. Furthermore, ASL showed remarkably few correlations to the other lignin methods, undermining the plausible assumption that an increase in acid soluble lignin would lead to a decrease in acid insoluble lignin. Such an inverse correlation was only found between ASL and ADL, and not for example between ASL and KL. This would suggest that these two traits can be investigated or selected for independently.
Extensive Variation among Accessions in the Release of Fermentable Sugars upon Mild Alkaline or Dilute Acid Pretreatment and Enzymatic Saccharification
In this comparative study, two pretreatment protocols were evaluated for determining the saccharification efficiency of ground stem samples. Samples were subjected to an alkaline pretreatment with NaOH and a dilute acid pretreatment with sulfuric acid. Saccharification efficiency was determined by the total release of fermentable sugars during pretreatment and subsequent enzymatic saccharification (Table 4). Glucose yields were similar between pretreatment methods, with average total glucose yields of 64.96 % for NaOH pretreated materials and 64.43 % for DA pretreated materials. Xylose yields, in contrast, were not similar between the two methods, and average total xylose yields were on average almost 13 % higher for NaOH pretreated materials (49.44 %) than for DA pretreated materials (36.82 %). These results show that at similar pretreatment severity, the enzymatic hydrolysis of xylose upon alkaline pretreatment (thus following partial removal of lignin) is more efficient than the combined chemical and enzymatic hydrolysis of xylose using sulfuric acid pretreatment.
At the alkaline pretreatment conditions evaluated in this study approximately 50 % of the lignin is likely to be removed, as predicted by a formula developed to estimate lignin removal during NaOH pretreatment [42]. During dilute acid pretreatment, however, the solubilization of lignin is only minimal [49]. Such a difference between pretreatment types in the amount of lignin remaining in pretreated sample is likely to affect the efficiency of enzymatic saccharification as lignin can irreversibly adsorb hydrolytic enzymes [9, 7]. In addition, since lignin and hemicellulosic polysaccharides are extensively cross-linked by ferulate bridges [4, 6, 50], the large difference in the amount of lignin that remains in the pretreated sample can possibly cause a difference in the release of xylose between the two pretreatments.
Both the alkaline and the dilute acid pretreatment resulted in significant differences among accessions for total glucose release (p= 0.007) (Tables 3 and 4). Significant differences among accessions for total xylose release, however, were only obtained using the alkaline pretreatment (p= 0.009) and not using the dilute acid pretreatment (p= 0.073), which can be attributed to the higher residual error in the data from the acid pretreated samples. However, both the alkaline and the dilute acid pretreatment consistently identified OPM-2 as the accession with the highest glucose (73 % for alkaline and 84 % for dilute acid) and xylose yields (55 % for alkaline and 41 % for dilute acid).
Even though mild pretreatment conditions were used, fairly good sugar release rates were obtained using this set of accessions. More importantly, however, a large variation in the performance of accessions at these mild conditions was observed. Especially for total glucose release from dilute acid pretreated materials, for which the best performing accession (OPM-2) yielded 33 % more glucose than the worst performing accession (OPM-9). This exemplifies the importance of feedstock optimization on process performance.
The two protocols were equally effective at screening differences in glucose release among accessions (p= 0.009), despite the larger range in the performance of accessions in glucose release upon acid pretreatment. This was mainly due to the larger residual error for glucose release using the acid pretreatment. As accessions could not be discriminated for xylose release using acid pretreatment, we conclude that in this study the alkaline pretreatment was overall more effective in screening variation in saccharification efficiency than the acid pretreatment. However, the type of pretreatment used for future screening purposes will largely depend on which pretreatment option will prevail in industry.
The Modified Klason Lignin Method Showed the Highest Potential for Predicting Saccharification Efficiency
Saccharification characters are generally strongly and negatively impacted by lignin content (Fig. 3). All different estimates of lignin content were significantly and negatively correlated to total glucose yields. Not all lignin estimates were, however, significantly correlated to xylose release after NaOH pretreatment. The only significant correlations to xylose yields of NaOH pretreated samples were found for ABL (r = −0.56) and ds-KL (r = −0.48). In contrast, correlations between glucose release and lignin concentrations were slightly stronger for NaOH pretreated (r = −0.74 to −0.85) than for DA pretreated samples (r = −0.63 to −0.84). These differences are most likely related to the different modes of action of the two pretreatment types. A major part of the lignin is likely to be removed upon NaOH pretreatment and fraction of the lignin that remains in the sample apparently doesn’t affect hydrolysis of hemicellulosic polysaccharides to a large extent. Also, in other studies, it was shown that glucose release is more negatively affected by residual lignin in the pretreated sample than xylose release [51].

Associations between saccharification characters and lignin estimates obtained using five different lignin determination methods. Regression lines and Pearson correlation coefficients are shown to indicate the strength and direction of trait correlations
Although all methods thus provided significant correlations to saccharification characters, there are differences observable when comparing the performance of the different lignin methods to predict the saccharification efficiency. First of all, it should be concluded that correlation patterns between saccharification efficiency and lignin content differed depending on the pretreatment method. The largest impact on sugar yields upon NaOH pretreatment was found for ds-KL. For DA pretreated samples, the largest impact on sugar yields were found for ADL. However, ds-KL also had a strong correlation to both xylose and glucose yields for DA pretreated samples. It was remarkable that the modified KL method performed considerably better than the reference as evaluated by the negative correlations with the different saccharification efficiency parameters. Apart from the fact that the smaller scale might be more suitable for this analysis, the higher correlation can also be due to a smaller technical error as more samples can be analyzed simultaneously.
The modified variant of the ADL method performed worse than the reference, as negative associations to saccharification characters were less strong for ADLseq than for ADL. Therefor, this method is not recommended, although sequential extraction of NDF, ADF, and ADL on the same bag provided a considerable reduction in time and labor.
Considering all lignin determination methods evaluated in this study, the strongest overall correlations to saccharification characters were found using the modified KL method. Lignin estimates obtained using this method were moderately correlated to xylose release (r = −0.48) when using NaOH pretreatment, but showed strong correlations to all other saccharification characters (r = −0.71 to −0.85). Hence, the lignin estimates obtained using this method had the highest predictive value for sample saccharification efficiency. Furthermore, the down-scaled Klason method can provide a large advantage over the unmodified Klason protocol, as it allows for more high-throughput analysis. This is especially true when additional modifications—such as disposable glass filters—are employed as proposed by Ibáñez and Bauer [48]. The modified Klason method furthermore has the additional advantage that it provides a reaction hydrolysate in which acid soluble lignin and neutral sugars can be determined. The latter is a considerable advantage as neutral sugar determination is essential to be able to express the release of fermentable sugars as a percentage of total available cell wall glucose and xylose. This method thus provides a complete analysis of biomass quality.
Conclusions
Large differences were observed between the various lignin methods. Despite that different lignin estimates were generally highly correlated to each other, they exhibited different correlation patterns to saccharification efficiency characters. The largest overall impact on saccharification yields was found for ds-KL. This modification of the KL method furthermore showed a large potential to discriminate differences among accessions and resulted in a reduction of time, labor, and technical error compared to the reference method. As it also allows for the parallel determination of neutral sugars and ASL, it provides a complete analysis of biomass quality.
References
Doblin MS, Pettolino F, Bacic A (2010) Plant cell walls: the skeleton of the plant world. Funct Plant Biol 37(5):357–381
Harris PJ, Stone BA (2008) Chemistry and Molecular Organization of Plant Cell Walls. In: Himmel ME (ed) Biomass Recalcitrance. Blackwell Publishing Ltd., Oxford, UK, pp 61–93
Rose JKC (2003) The plant cell wall. Annual Plant Reviews, vol 8. CRC Press and WileyBlackwell, Oxford
Carpita NC (1996) Structure and biogenesis of the cell walls of grasses. Annu Rev Plant Physiol Plant Mol Biol 47(1):445–476. doi:10.1146/annurev.arplant.47.1.445
Grabber JH (2005) How do lignin composition, structure, and cross-linking affect degradability? A review of cell wall model studies. Crop Sci 45(3):820–831. doi:10.2135/cropsci2004.0191
Grabber JH, Ralph J, Lapierre C, Barrière Y (2004) Genetic and molecular basis of grass cell-wall degradability. I. Lignin–cell wall matrix interactions. C R Biol 327(5):455–465. doi:10.1016/j.crvi.2004.02.009
Zhao X, Zhang L, Liu D (2012) Biomass recalcitrance. Part I: the chemical compositions and physical structures affecting the enzymatic hydrolysis of lignocellulose. Biofuels Bioprod Biorefin 6(4):465–482. doi:10.1002/bbb.1331
Himmel ME, Picataggio SK (2008) Our challenge is to acquire deeper understanding of biomass recalcitrance and conversion. In: Himmel ME (ed) Biomass Recalcitrance. Blackwell Publishing Ltd., Oxford, UK, pp 1–6
Jørgensen H, Kristensen JB, Felby C (2007) Enzymatic conversion of lignocellulose into fermentable sugars: challenges and opportunities. Biofuels Bioprod Biorefin 1(2):119–134. doi:10.1002/bbb.4
Wyman CE (2007) What is (and is not) vital to advancing cellulosic ethanol. Trends Biotechnol 25(4):153–157. doi:10.1016/j.tibtech.2007.02.009
Simmons BA, Loqué D, Ralph J (2010) Advances in modifying lignin for enhanced biofuel production. Curr Opin Plant Biol 13(3):312–319. doi:10.1016/j.pbi.2010.03.001
Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Ralph SA, Christensen JH (2004) Lignins: Natural polymers from oxidative coupling of 4-hydroxyphenyl-propanoids. Phytochem Rev 3(1–2):29–60
Vanholme R, Demedts B, Morreel K, Ralph J, Boerjan W (2010) Lignin biosynthesis and structure. Plant Physiol 153(3):895–905
Frei M (2013) Lignin: characterization of a multifaceted crop component. The Scientific World Journal 2013
Davin LB, Patten AM, Jourdes M, Lewis NG (2008) Lignins: a twenty-first century challenge. In: Himmel ME (ed) Biomass Recalcitrance. Blackwell Publishing Ltd., Oxford, UK, pp 213–305
Hatfield R, Fukushima RS (2005) Can lignin be accurately measured? Crop Sci 45(3):832–839. doi:10.2135/cropsci2004.0238
Vermerris W, Nicholson R (2006) Isolation and Identification of Phenolic Compounds. Phenolic Compound Biochemistry. Springer Netherlands, pp 151–196. doi:10.1007/978-1-4020-5164-7_4
Browning BL (1967) Methods of wood chemistry. Volumes I & II. Methods of wood chemistry Volumes I & II
Goering HK, Van Soest PJ (1970) Forage fiber analyses (apparatus, reagents, procedures, and some applications). Agric. Handbook No. 379. U.S. Agricultural Research Service, Washington, DC
Van Soest PJ (1963) Use of detergents in analysis of fibrous feeds. II. A rapid method for the determination of fiber and lignin. J Assoc Off Anal Chem 46:829–835
Van Soest PJ (1967) Development of a comprehensive system of feed analyses and its application to forages. J Anim Sci 26(1):119–128
Kondo T, Mizuno K, Kato T (1987) Some characteristics of forage plant lignin. Japan Agric Res Q 21(1):47–52
Hatfield RD, Jung H-JG, Ralph J, Buxton DR, Weimer PJ (1994) A comparison of the insoluble residues produced by the Klason lignin and acid detergent lignin procedures. J Sci Food Agric 65(1):51–58. doi:10.1002/jsfa.2740650109
Johnson DB, Moore W, Zank L (1961) The spectrophotometric determination of lignin in small wood samples. Tappi 44(11):793–798
Fukushima RS, Hatfield RD (2001) Extraction and isolation of lignin for utilization as a standard to determine lignin concentration using the acetyl bromide spectrophotometric method (journal of agricultural and food chemistry). J Agric Food Chem 49(7):3133–3139. doi:10.1021/jf010449r
Moreira-Vilar FC, Siqueira-Soares RdC, Finger-Teixeira A, Oliveira DMd, Ferro AP, da Rocha GJ, Ferrarese MdLL, dos Santos WD, Ferrarese-Filho O (2014) The Acetyl Bromide Method Is Faster, Simpler and Presents Best Recovery of Lignin in Different Herbaceous Tissues than Klason and Thioglycolic Acid Methods. PLoS ONE 9 (10)
Goff BM, Murphy PT, Moore KJ (2012) Comparison of common lignin methods and modifications on forage and lignocellulosic biomass materials. J Sci Food Agric 92(4):751–758
Lacerda RS, Gomide CA, Fukushima RS, Herling VR (2006) Lignin concentration in oat (Avena byzantina L.) aerial part as measured by four analytical methods. Braz J Vet Res Anim Sci 43:400–407
Fukushima RS, Hatfield RD (2004) Comparison of the acetyl bromide spectrophotometric method with other analytical lignin methods for determining lignin concentration in forage samples (Journal of Agricultural and Food Chemistry). J Agric Food Chem 52(12):3713–3720. doi:10.1021/jf035497l
Takahashi T, Fujii T, Yasuoka Y (2004) Estimation and comparison of acid detergent lignin and acetyl bromide lignin in fallen leaves using near-infrared spectroscopy. Int J Remote Sens 25(24):5585–5600. doi:10.1080/01431160410001735102
Brinkmann K, Blaschke L, Polle A (2002) Comparison of different methods for lignin determination as a basis for calibration of near-infrared reflectance spectroscopy and implications of lignoproteins. J Chem Ecol 28(12):2483–2501
Jung H-JG, Varel VH, Weimer PJ, Ralph J (1999) Accuracy of Klason lignin and acid detergent lignin methods as assessed by bomb calorimetry. J Agric Food Chem 47(5):2005–2008
Van Soest P, Wine R (1967) Use of detergents in the analysis of fibrous feeds. IV. Determination of plant cell-wall constituents. J Assoc Off Anal Chem 50(1):50–55
Fukushima RS, Kerley MS (2011) Use of lignin extracted from different plant sources as standards in the spectrophotometric acetyl bromide lignin method. J Agric Food Chem 59(8):3505–3509. doi:10.1021/jf104826n
Lygin AV, Upton J, Dohleman FG, Juvik J, Zabotina OA, Widholm JM, Lozovaya VV (2011) Composition of cell wall phenolics and polysaccharides of the potential bioenergy crop –Miscanthus. GCB Bioenergy 3(4):333–345. doi:10.1111/j.1757-1707.2011.01091.x
Sluiter A, Hames B, Ruiz R, Scarlata C, Sluiter J, Templeton D, Crocker D (2012) Determination of Structural Carbohydrates and Lignin in Biomass. Laboratory Analytical Procedure. National Renewable Energy Laboratory, Golden, CO
Lin SY, Dence CW (1992) The Determination of Lignin. Methods in Lignin Chemistry. Springer Series in Wood Science. Springer, Berlin, pp 33–61. doi:10.1007/978-3-642-74065-7_3
Xu N, Zhang W, Ren S, Liu F, Zhao C, Liao H, Xu Z, Huang J, Li Q, Tu Y, Yu B, Wang Y, Jiang J, Qin J, Peng L (2012) Hemicelluloses negatively affect lignocellulose crystallinity for high biomass digestibility under NaOH and H2SO4 pretreatments in Miscanthus. Biotechnol Biofuels 5(1):58
Torres A, Noordam-Boot CM, Dolstra O, van der Weijde T, Combes E, Dufour P, Vlaswinkel L, Visser RF, Trindade L (2014) Cell Wall Diversity in Forage Maize: Genetic Complexity and Bioenergy Potential. BioEnergy Research:1–16. doi:10.1007/s12155-014-9507-8
Torres AF, Weijde T, Dolstra O, Visser RGF, Trindade LM (2013) Effect of Maize Biomass Composition on the Optimization of Dilute-Acid Pretreatments and Enzymatic Saccharification (BioEnergy Research). Bioenergy Research:1–14. doi:10.1007/s12155-013-9337-0
Pedersen M, Meyer AS (2010) Lignocellulose pretreatment severity–relating pH to biomatrix opening. New Biotechnol 27(6):739–750
Silverstein RA, Chen Y, Sharma-Shivappa RR, Boyette MD, Osborne J (2007) A comparison of chemical pretreatment methods for improving saccharification of cotton stalks. Bioresour Technol 98(16):3000–3011. doi:10.1016/j.biortech.2006.10.022
Selig M, Weiss N, Ji Y (2008) Enzymatic saccharification of lignocellulosic biomass. Laboratory Analytical Procedure. National. Renewable Energy Laboratory, Golden, CO
Heaton EA, Dohleman FG, Miguez AF, Juvik JA, Lozovaya V, Widholm J, Zabotina OA, McIsaac GF, David MB, Voigt TB, Boersma NN, Long SP (2010) Miscanthus. A promising biomass crop. Adv Bot Res 56:76–137
Robson P, Mos M, Clifton-Brown J, Donnison I (2012) Phenotypic variation in senescence in miscanthus: Towards optimising biomass quality and quantity. Bio Energy Res 5(1):95–105. doi:10.1007/s12155-011-9118-6
Hodgson EM, Lister SJ, Bridgwater AV, Clifton-Brown J, Donnison IS (2010) Genotypic and environmentally derived variation in the cell wall composition of Miscanthus in relation to its use as a biomass feedstock. Biomass Bioenergy 34(5):652–660. doi:10.1016/j.biombioe.2010.01.008
Jung HG, Mertens DR, Payne AJ (1997) Correlation of acid detergent lignin and klason lignin with digestibility of forage dry matter and neutral detergent fiber. J Dairy Sci 80(8):1622–1628. doi:10.3168/jds.S0022-0302(97)76093-4
Ibáñez AB, Bauer S (2014) Downscaled method using glass microfiber filters for the determination of Klason lignin and structural carbohydrates. Biomass Bioenergy 68:75–81. doi:10.1016/j.biombioe.2014.06.013
Yang B, Wyman CE (2008) Pretreatment: the key to unlocking low‐cost cellulosic ethanol. Biofuels Bioprod Biorefin 2(1):26–40
Hatfield RD, Ralph J, Grabber JH (1999) Cell wall cross-linking by ferulates and diferulates in grasses1. J Sci Food Agric 79:403–407
Chen M, Zhao J, Xia L (2009) Comparison of four different chemical pretreatments of corn stover for enhancing enzymatic digestibility. Biomass Bioenergy 33(10):1381–1385
Acknowledgments
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement n° 289159. The genotypes used in this study originate from a field trial established in Wageningen using 15 genotypes selected from germplasm collections of the miscanthus breeding programs of Wageningen University and Aberysthwyth University. We gratefully acknowledge our partners in this project: Aberystwyth University for the supply of a part of the genotypes and Kai-Uwe Schwartz and Heike Meyer for the micro-propagation of the planting material. We further acknowledge Genencor International B.V./DuPont Industrial Biosciences for kindly supplying us with their commercial Accellerase 1500 enzyme cocktail used in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van der Weijde, T., Torres, A.F., Dolstra, O. et al. Impact of Different Lignin Fractions on Saccharification Efficiency in Diverse Species of the Bioenergy Crop Miscanthus. Bioenerg. Res. 9, 146–156 (2016). https://doi.org/10.1007/s12155-015-9669-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12155-015-9669-z