
Abstract
Alcohols of increasing alkyl chain length were investigated as precipitants in an ionic liquid (IL) pretreatment system. Switchgrass samples pretreated by 1-ethyl-3-methylimidazolium acetate were characterized after the use of different alkyl chain lengths of alcohols as antisolvents. The resulting IL-pretreated switchgrass (PSG) samples were characterized by enzymatic hydrolysis, cross polarization/magic angle spinning (CP/MAS) 13C nuclear magnetic resonance (NMR), Fourier transform infrared spectroscopy (FTIR), and 2D NMR spectroscopy. Glucan digestibilities of PSG samples were ∼80 % after 72 h at 5 mg protein g−1 glucan regardless of the antisolvent used. The use of 1-octanol as an antisolvent, with 10 % water to allow for use of wet biomass, enabled a partial lignin fractionation and multiphase separation for the IL recycle without compromising the chemical structure of the carbohydrates and lignin from the PSG. Lignin fragments were observed in the IL after pretreatment by gel permeation chromatography (GPC). After separation, both the IL and the octanol antisolvent were reused for switchgrass pretreatment and precipitation for an additional 3 cycles. The PSG samples derived from recycled IL were rapidly hydrolyzed, and a high glucan digestibility of 80 % was obtained even at a low enzyme loading of 5 mg protein g−1 glucan. 2D NMR analysis of residual solids of PSG post-enzymatic hydrolysis revealed that lignin in these residual solids was depolymerized. This strategy enables an ease in separation of pretreated lignocellulosic solids, reduced water use, and recycle of both IL and the antisolvent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The use of nonfood lignocellulosic biomass as a renewable and sustainable energy source is one of a suite of technologies which must be employed if a carbon neutral economy or significant carbon reduction to the environment is to be achieved. Developing a biofuel infrastructure is especially important for air and freight where other technologies will not be employed in the near term. Recalcitrance of nonfood lignocellulose is one of the major obstacles to effective production of biofuels and biobased products. The recalcitrant nature of lignocellulosic biomass is attributed to many factors, such as low substrate accessibility, high degree of polymerization of cellulose, the presence of lignin and hemicellulose, high crystallinity, large particle size, and low porosity [17]. Consequently, lignocellulosic biomass must be pretreated in order to overcome the recalcitrant nature of lignocellulose and also achieve lignin separation for downstream upgrading to value-added products. The separation of lignin and its subsequent valorization are necessary steps in the conversion of the cellulosic component of the biomass to biofuels. To that end, several pretreatment approaches have been proposed, namely, dilute acid (DA), ammonia fiber expansion (AFEX), soaking in aqueous ammonia (SAA), steam explosion, and lime pretreatments [4, 7]. Many of these pretreatments are ineffective at increasing cellulose surface accessibility to cellulase and require high cellulase loading to achieve high sugar yields [11, 18].
Many ionic liquids (ILs) have shown to be very high performing solvents, having been shown to decrease the degree of crystallinity of cellulose, increase cellulose accessibility, and both dissolve and depolymerize lignin, depending on the type of ILs [1, 5]. However, the excellent lignin extraction and solvation properties of certain ILs necessitate the subsequent recovery of the solutes. In addition, as with any solvent in an industrial process, an IL pretreatment process will require recovery and reuse of the IL. The very properties which make ILs excellent solvents (i.e., thermal and chemical stability and excellent solvation properties) require new systems to be developed to remove dissolved species and recycle the IL. Systems such as distillation cannot be as readily used with thermally stable ILs as with low-boiling-point organic solvents. Therefore, straightforward solid-liquid separation systems have been proposed where, typically, water is employed as a precipitant for both the lignin and cellulose component. However, this solute removal system subsequently impacts the IL recycle unit operations, as the water needs to be removed from the IL. The use of water can impose both an energy, environmental, and performance penalty; in this study, alcohols were investigated as alternative precipitating agents for the IL pretreatment process.
Herein, we investigate the effect of different chain lengths of alcohols and IL pretreatment using 1-ethyl-3-methylimidazolium acetate ([C2C1Im][OAc]) on lignin extraction efficacy and recycle of IL. The spent IL was reused for four times to pretreat switchgrass. Pretreatment efficiency of recycled IL was probed by enzymatic hydrolysis and cross polarization magic-angle spinning (CP/MAS) 13C NMR of pretreated biomass by recycled IL. 13C-1H heteronuclear single-quantum coherence (HSQC) NMR was employed to probe changes in lignin interunit linkages and relative ratios of aromatic units on residual solids after enzymatic hydrolysis.
Materials and Methods
Chemicals and Materials
All chemicals were of reagent grade and purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise noted. [C2C1Im][OAc] was purchased from BASF (98 % purity, lot no. 11-0005) and used as received. The cellulase (CTec 2) and hemicellulase (HTec 2) mixtures were provided as a gift by Novozymes North America (Franklinton, NC, USA), containing 188 and 186 mg protein mL−1, respectively. Alamo switchgrass (Panicum virgatum L.) was provided by Dr. Daniel Putnam, University of California at Davis. Switchgrass was milled by a Wiley Mill through a 2-mm screen and separated by a vibratory sieve system (Endecotts, Ponte Vedra, FL, USA).
IL Pretreatment
IL pretreatment of lignocellulose was conducted. Figure 1 represents a schematic diagram of the IL pretreatment process, followed by enzymatic hydrolysis used in the present study. Briefly, 15 % (w/w) switchgrass in [C2C1Im][OAc] was loaded in a Syrris globe reactor at 140 °C for 1.5 h, unless otherwise noted. The hydrogel-like solution was allowed to cool to 50 °C, and two volumes of antisolvents, described below, were added to solubilize partial lignin and precipitate dissolved switchgrass (Fig. 1, precipitation tank). L2 (Fig. 1) denotes the lignin solubilized in the IL. After centrifugation, the supernatant was collected (Fig. 1, decanter). An additional one volume of antisolvent was then used to wash solubilized lignin from the pretreated switchgrass. This lignin stream is denoted in Fig. 1 as L3. After centrifugation, the pellets were washed by one volume of deionized water twice to remove residual [C2C1Im][OAc] and antisolvents from the solid pellets. The resulting solid pellet pretreated switchgrasses (PSGs) were used in the enzymatic hydrolysis experiments. The octanol-IL-mixture-containing solubilized lignin was allowed to settle; after 24 h any solids that precipitated could be removed. The two-phase system was separated via careful pipetting thereby allowing the reuse of both the IL and the antisolvent.

Lignocellulose chemical structure was disrupted by ionic liquids and dissolved lignocellulose was regenerated in an antisolvent. Partial lignin from lignocellulose can be fractionated from a mixture of ionic liquid and antisolvent, enabling fractionation of lignin for downstream conversion to value-added products. The mixture of ionic liquid and antisolvent can be separated and recycled. Regenerated lignocellulose was then hydrolyzed by cellulolytic enzymes to release sugars for downstream conversion to biofuels and/or biochemicals
Influence of Alkyl Chain Length of Alcohols on Enzymatic Hydrolysis Characteristics of PSG
Alcohols of increasing alkyl chain length were used in this study: methanol, ethanol, 1-propanol, 1-butanol, 1-hexanol, 1-octanol, and isopropanol. In addition, experiments with acetone, acetone-water (1:1), and water whose behavior is reported in the literature were conducted as controls. These results allow us to investigate the influence of alkyl length on lignin extraction and enzymatic hydrolysis efficiency.
Carbohydrate and Lignin Assays
The carbohydrate composition of lignocellulose and residual pretreated lignocellulose after hydrolysis was determined with a modified quantitative saccharification (QS) procedure [8]. In the modified QS, secondary hydrolysis was conducted in the presence of 1 % (w/w) sulfuric acid at 121 °C for 1 h to more accurately determine the quantities of sugars susceptible to acid degradation (e.g., xylan). Monomeric sugars in the supernatant were measured with an Agilent HPLC equipped with a Bio-Rad Aminex HPX-87H column (Richmond, CA, USA) at a rate of 0.6 mL of 0.1 % (v/v) sulfuric acid per min at 60 °C. The standard NREL biomass protocol was used to measure lignin and ash [14]. Briefly, solids remaining after two-stage acid hydrolysis were held at 105 °C overnight. The mass of the dried solids corresponds to the amount of acid-insoluble lignin and ash in the sample. The mass of the ash-only fraction was then determined gravimetrically via combustion by heating the solids to 575 °C for 24 h. Percent acid-soluble lignin in the sample was determined by measuring the UV absorption of the acid hydrolysis supernatant at 240 nm. All carbohydrate and lignin assays were conducted in triplicate.
Enzymatic Hydrolysis
The pretreated samples were diluted to 100 g solid L−1 in a 50-mM sodium citrate buffer (pH 4.8) supplemented with 0.1 % (w/v) NaN3, which prevented the growth of microorganisms. All enzymatic hydrolysis experiments were conducted in triplicate. Pretreated samples were completely suspended in a rotary shaker at 250 rpm at 50 °C. The enzyme loadings were kept constant at 20 and 5 mg protein per gram of glucan (initial glucan) using commercial CTec2 and HTec2 (9:1 CTec2/HTec2 by weight). Eight hundred microliters of well-mixed hydrolysate was removed, followed by immediate centrifugation at 13,000 rpm for 5 min. Exactly 500 μL of the supernatant was transferred to another microcentrifuge tube and stayed at room temperature for 30 min, to allow the conversion of all cellobiose to glucose. The supernatant was then acidified by adding 30 μL of 10 % (w/w) sulfuric acid, followed by freezing overnight. The frozen samples were thawed, mixed well, and then centrifuged at 13,000 rpm for 5 min, to remove any precipitated solid sediments. The soluble glucose and xylose in the enzymatic hydrolysate were measured by HPLC equipped with a Bio-Rad HPX-87H column at a rate of 0.6 mL of 0.1 % (v/v) sulfuric acid per min at 60 °C. Galactose and mannose co-eluted with xylose. After 72-h hydrolysis, the remaining hydrolysate was transferred to a 50-mL centrifuge tube and centrifuged at 4500 rpm for 15 min, and soluble sugar content was determined using the same procedure as other hydrolysate samples, as described above. After all remaining hydrolysate was decanted, the pellets were resuspended in 30 mL of water and centrifuged to remove residual soluble sugars from the pellets. The sugar content of the washed pellets was determined by modified QS as described above. Enzymatic glucan digestibility after 72 h was calculated using the ratio of soluble glucose in the supernatant to the sum of this soluble glucose and the glucose equivalent of the residual glucan.
Isolation of Enzymatic Mild Acidolysis Lignin (EMAL)
Ball milling of biomass was performed using a Retsch PM 100 planetary ball mill spinning at 600 rpm with zirconium dioxide (ZrO2) container and balls. The ball milling conditions are described elsewhere [3]. Briefly, the ball-milled biomass samples were treated with cellulase (CTec2) and hemicellulase (HTec2) in the amount of 50 mg protein g−1 biomass. The enzymatic hydrolysis was carried out at 50 °C for 48 h at 2 % consistency in the presence of 2 % Tween 20 in 50-mM citrate buffer (pH ∼4.8). The insoluble materials were washed with deionized water, and a fresh batch of enzymes, in the same quantity, was added for another 48 h. The insoluble materials remaining after enzymatic hydrolysis were washed with deionized water to remove soluble sugars. Residual proteins on the surface of solid pellets were then washed twice with 6 M guanidine hydrochloride (Gnd HCl) and freeze-dried. The crude lignin obtained was further subjected to mild acid hydrolysis using an azeotrope of dioxane-water (96:4 (v/v)) containing 0.01 N HCl under nitrogen atmosphere. The resulting suspension was centrifuged, and the supernatant was collected. The supernatant was neutralized with 2 M sodium bicarbonate and then added drop-wise into 1 L acidified water (pH 2.0). The precipitated lignin was allowed to equilibrate overnight, recovered by centrifugation, washed with deionized water twice, and freeze-dried.
Gel Permeation Chromatography (GPC)
Lignin solution, 1 % (w/v) EMAL of switchgrass, was prepared in analytical-grade 1-methyl-2-pyrrolidinone (NMP). Streams L2 and L3 were analyzed by taking a minute amount of L2 and L3 aliquots in 200 μL NMP. EMAL produced from switchgrass was also evaluated as the control. The polydispersity of dissolved lignin was determined using analytical techniques involving GPC UV-A absorbance (GPC UV-A290) as previously described [1]. An Agilent 1200 series binary LC system (G1312B) equipped with DA (G1315D) detector was used. Separation was achieved with a Mixed-D column (5-μm particle size, 300 × 7.5-mm id, linear molecular mass range of 200 to 400,000 μ, Polymer Laboratories) at 80 °C using a mobile phase of NMP at a flow rate of 0.5 mL min−1. Absorbance of materials eluting from the column was detected at 290 nm (UV-A). Intensities were area-normalized and molecular mass estimates were determined after calibration of the system with polystyrene standards. Based on current knowledge, polystyrene does not represent the geometry, or chemistry of the lignin molecule, but is the currently used standard for GPC calibration in the literature. Polystyrene calibrations were conducted here to confirm correct GPC system behavior and so that the data presented in this study may be compared to other published data using similar GPC systems and methods.
2D 13C-1H HSQC NMR Spectroscopy
Residual solids after enzymatic hydrolysis were ball-milled as previously described [3, 6]. The gels were formed using dimethyl sulfoxide (DMSO)-d6 and pyridine-d5 and sonicated until homogenous in a Branson 2510 tabletop cleaner (Branson Ultrasonic Corporation, Danbury, CT, USA). The temperature of the bath was closely monitored and maintained below 55 °C. The homogeneous solutions were transferred to NMR tubes. HSQC spectra were acquired at 25 °C using a Bruker Avance 600-MHz instrument equipped with a 5-mm inverse-gradient 1H/13C cryoprobe using a q_hsqcetgp pulse program (ns = 200, ds = 16, number of increments = 256, d 1 = 1.0 s) [2]. Chemical shifts were referenced to the central DMSO peak (δ C/δ H 39.5/2.5 ppm). Assignment of the HSQC spectra is described elsewhere [3, 16]. A semiquantitative analysis of the volume integrals of the HSQC correlation peaks was performed using Bruker’s Topspin 3.1 (Windows) processing software. A Gaussian apodization in F2 (LB = −0.50, GB = 0.001) and squared cosine-bell in F1 (LB = −0.10, GB = 0.001) were applied prior to 2D Fourier transformation.
Solid-State NMR (ssNMR)
The CP/MAS 13C-NMR spectra of all samples were obtained on a Bruker Avance I 500-MHz NMR spectrometer operating at the resonance frequencies of 500.23 MHz for 1H and 125.80 MHz for 13C, using a double-resonance Bruker 4.0-mm broad-band CP-MAS probe spinning at 13–14 kHz. Cross polarization for 2-ms contact time was achieved using a 1H 90° pulse width of 4.2 μs at 60-kHz two-pulse phase-modulated proton decoupling field and 2-s recycle delay. Total accumulation time was between 1000 and 3000 transients. All spectra were collected at room temperature and referenced against the chemical shifts of adamantane at 38.48 and 29.45 ppm. According to the C4 peak-deconvolution method [3], the degree of crystallinity was determined and expressed as crystallinity index (CrI). The CrI value was calculated from the ratio of the crystalline area over the total area, where separation of crystalline (δ 86–92 ppm) and amorphous (δ 79–86 ppm) fractions was based on Gaussian line shape function.
Fourier Transform Infrared Spectroscopy (FTIR)
All FTIR spectra were collected on the Thermo Nicolet 8700 spectrometer equipped with attenuated total reflectance (ATR) mode (Thermo Fisher Scientific, Inc., USA). Sixty scans at resolution of 4 cm−1 were averaged for each sample. A background was collected prior to analyzing each sample and subtracted from each spectrum. Spectra width is 4000–600 cm−1. All the spectra are auto-baseline-corrected using the Omnic software.
Results and Discussion
Influence of Alcohol Alkyl Chain Length on PSG Characteristics and Their Enzymatic Hydrolysis Efficiency
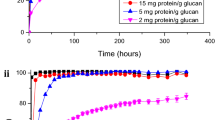
Switchgrass samples of 2-mm particle size were pretreated by [C2C1Im][OAc]. After IL pretreatment, an antisolvent was added to precipitate dissolved switchgrass samples. A number of alcohols of different alkyl chain lengths were evaluated as antisolvents—methanol, ethanol, 1-propanol, 1-butnaol, 1-hexanol, and 1-octanol. Water, 2-propanol, acetone, and acetone-water (50/50) were also used for comparison as these antisolvents were previously reported. Importantly, an increase in hydrophobicity of alcohols was observed as a function of chain length, which for octanol led to a two-phase IL-alcohol system. PSG samples from different antisolvents were hydrolyzed by CTec2 and HTec2 at 5 and 20 mg protein g−1 glucan. The nitrogen content in the PSG was tested to evaluate protein binding to all the biomass tested, and this was found to be negligible (below instrument limits of quantification). At a low enzyme loading of 5 mg protein g−1 glucan, enzymatic glucan digestibility yields of PSG were higher than 78 %, regardless of antisolvents used (Fig. 2a). An increase in enzyme loading to 20 mg protein g−1 glucan (Fig. 2b) showed a faster initial hydrolysis rate, as reflected in more than twofold increase in glucan digestibilities within a 1-h hydrolysis time. All enzymatic hydrolysis reactions at 20 mg protein g−1 glucan yielded higher than 90 % glucan digestibility, regardless of the choice of an antisolvent. These results suggested the high biomass pretreatment efficiency by IL under the current pretreatment condition, as shown in high glucan digestibilities of PSG even at a fourfold decrease in enzyme loading.

Enzymatic hydrolysis profiles of [C2C1Im][OAc]-pretreated switchgrass samples using different alcohols as antisolvents at 5 mg protein g−1 glucan (a) show high glucan digestibilities and using different antisolvents do not show statistically significant difference in glucan digestibilities. An increase in enzyme loading to 20 mg protein g−1 glucan (b) to hydrolyze pretreated switchgrass yielded faster initial hydrolysis rates (1-h hydrolysis time) and slightly higher glucan digestibilities
Influence of Alcohol Alkyl Chain Length on Lignin Elution Characteristics After IL Pretreatment
Most current pretreatment studies focus on lowering the degree of lignocellulose recalcitrance to enhance enzymatic glucan digestibility. Herein, we investigated how lignin biopolymers behave after IL pretreatment. As shown in Fig. 1, lignin from lignocellulose could partition into IL, as IL ([C2C1Im][OAc] in this study) has been shown to be effective not only at pretreating lignocellulose but also at partially solubilizing lignin [12]. Varanasi et al. [15] showed that during IL pretreatment, a part of lignin within lignocellulose was hydrolyzed and some small lignin fragments were detected in IL. As such, we hypothesized that although the choice of antisolvents would not affect enzymatic hydrolysis performance of PSG, different antisolvents could influence lignin elution profiles of recovered lignin, with certain lignin fragments preferentially partitioning into the alcohol phase.
Molecular weight distribution (MWD) of lignin in different antisolvents was evaluated by GPC. EMAL of SG was used as a control and the GPC chromatogram of EMAL of SG showed a bi-modal distribution (Fig. 3), suggesting that EMAL of SG (stream L1, Fig. 1) consists of mainly high-molecular-weight lignin fragments with a trace amount of low-molecular-weight lignin fragments. After IL pretreatment (stream L2, Fig. 1), partial lignin was hydrolyzed as shown in polydispersed characteristic of L2 (Fig. 3). Addition of the antisolvents in the pretreatment slurry (stream L3, Fig. 1) showed similar molecular weight distribution as L2, which was because most of the antisolvents in this study are miscible with IL except 1-hexanol and 1-octanol (partially miscible). Interestingly, in the case of L3 of 1-octanol, a strong signal was observed in both excluded and retained regions, implying that both high and low-molecular-weight lignin fragments migrated to octanol, which indicates a more complete removal of the lignin from the IL.

GPC elution profiles of extracted lignin in [C2C1Im][OAc] before and after addition of different antisolvents revealed lignin depolymerization during pretreatment
Enzymatic Hydrolysis Efficiency of PSG by Recycled IL
One of the major challenges in IL pretreatment is the recycle of ionic liquids. IL recycle is a difficult task, as most antisolvents are miscible with IL. We found that for IL-octanol solution after pretreatment, adding 10 % water created a meniscus between octanol and IL, enabling a multiphase separation and IL recycle. Consequently, octanol and IL phases were reused for three subsequent IL pretreatments. Octanol was observed to be darker in color following each recycle, which might be due to migration of lignin fragments into the octanol phase, corroborated by the presence of a strong signal of lignin elution profiles as detected by GPC. PSG samples by the first, second, and third recycled IL were enzymatically hydrolyzed, and the enzymatic saccharification efficiency values were similar to that of fresh IL (Fig. 4), suggesting good recycling efficiency of the IL-octanol system.

Recycled [C2C1Im][OAc] was used to pretreat switchgrass for three times, and no decrease in the glucan digestibility of pretreated switchgrass was observed
Structural Characteristics of PSG by Recycled IL via Solid-State NMR and FTIR
CP/MAS 13C NMR spectroscopy is a useful tool to analyze breaking and rearranging of hydrogen bonding of cellulose chains. C4 and C6 regions of CP/MAS 13C NMR spectra of lignocellulose reveal the state of cellulose crystallinity and hydrogen bonding among cellulose chains, respectively [9, 10]. C4 peak deconvolution was applied to separate the contribution of crystalline and amorphous cellulose on intact SG sample (Fig. 5b) and CrI was calculated as previously described [13]. CrI of intact SG was found to be ∼38 %, which is in agreement with the previous reported value [13], suggesting a robustness of this CrI calculation method. The PSG samples by fresh and recycled IL were probed by CP/MAS 13C NMR. PSGs were lyophilized prior to CP/MAS 13C NMR analysis. It should be noted that we do not see changes in CrI after sample lyophilization. CrI of PSG samples was 15.9, 12.1, and 14.0 % for PSG by fresh and the first and second (round) recycled IL, respectively. Moreover, the peak shift in the C6 region (Fig. 5c) to the higher field after pretreatment by the recycled IL implied that both fresh and recycled IL could effectively disrupt hydrogen bonding among cellulose chains of switchgrass. These results suggest that recycled IL from the [C2C1Im][OAc]-octanol system still retains pretreatment efficacy similar to the fresh IL.

Full CP/MAS 13C NMR spectra (δ 120–50, a), C4 region (δ 94–80, b), and C6 region (δ68–58, c) of intact SG and PSG by fresh and recycled [C2C1Im][OAc] show that recycled [C2C1Im][OAc] can effectively pretreat biomass. Fingerprint region of FTIR of intact SG and PSG by fresh and recycled IL (d)
Changes in the FTIR spectra of PSG by fresh IL compared to that of intact SG were observed in the fingerprint region (Fig. 5d). Full FTIR spectra of intact SG and PSG by fresh IL and the first and second recycled IL are shown in Fig. S2 (Supporting Material). A significant decrease in the spectral intensity of etheric Caromatic–O–Caliphatic at 1249 cm−1 of PSG indicated that pretreatment severed the lignin and carbohydrate bonds, releasing lignin fragments in the [C2C1Im][OAc] after pretreatment as shown in GPC results (Fig. 3). A decrease in the spectral intensity at 1730 cm−1 (υ(C═O), carbonyl stretching), suggesting that acetylated groups from hemicelluloses were removed as a result of IL pretreatment. “’Moreover, enhanced band intensities at 897 (υ as(ring), anomeric vibration at β-glycosidic linkages) and 1159 cm−1 (υ as(COC), COC antisymmetric stretching) of PSG by fresh and recycled [C2C1Im][OAc] compared to that of intact SG, suggesting that PSG samples were more disordered (amorphous) and [C2C1Im][OAc] could be reused (three times in the present study) without reducing its pretreatment efficiency.
Chemical Structure Characteristics of Lignin from PSG Post-enzymatic Hydrolysis via 2D NMR
13C-1H HSQC NMR was performed to investigate changes of aliphatic (lignin side chain units, Fig. 6a, b) and aromatic (Fig. 6c, d) regions of intact SG and PSG samples post-enzymatic hydrolysis by fresh and recycled IL. Aliphatic regions of intact SG exhibited two distinct peaks of 2-O-Ac-β-D-Xylp(R) (X′2) and 3-O-Ac-β-D-Xylp(R) (X′3), suggesting acetylation of hemicelluloses in the intact SG. Moreover, lignin of intact SG consists mainly of β-aryl ether (β-O-4), resinol (β-5), and dibenzodioxocins. The β-aryl ether linkage is the most abundant in plant lignins and can be easily cleaved compared to resinol and phenylcoumaran. After IL pretreatment, the β-aryl ether cross peak became weaker, suggesting that the ether bonds were severed and lignin fragments were possibly released. This result is in agreement with our GPC elution profiles, revealing different lignin fragments released in the [C2C1Im][OAc] after pretreatment (Fig. 3). Moreover, dibenzodioxocins were not observed in PSG samples. Dibenzodioxocins are believed to be branching points of lignin. Hence, an absence of dibenzodioxocins after pretreatment suggested that lignin was depolymerized during pretreatment, corroborating with the presence of lignin fragments in the L2 and L3 stream by GPC (Fig. 3). Moreover, X′2 and X′3 disappeared after IL pretreatment and subsequent enzymatic hydrolysis, suggesting that a large amount of hemicelluloses were removed after enzymatic hydrolysis.

13C-1H HSQC NMR spectra of residual solids post-enzymatic hydrolysis of PSG by fresh IL show lignin structural integrity of residual solids
Examination of the aromatic region (Fig. 6c, d) demonstrated that PSG did not show any alteration in the signals for major aromatic units compared to that of intact SG except the disappearance of the ferulic acid (FAα) and para-coumaric acid (pCAα) after IL pretreatment. FA and pCA are believed to bridge between lignin and carbohydrate units as lignin-carbohydrate complex (LCC) linkages. Hence, post-enzymatic hydrolysis, a part of FA and pCA might become labile in the enzymatic hydrolysis solution. These results suggest that while IL pretreatment could hydrolyze some lignin into smaller fragments, the remaining lignin in PSG samples has similar aromatic structures to lignin of intact SG. Similar results were observed on lignin post-enzymatic hydrolysis from PSG by the first and second recycled [C2C1Im][OAc] (Fig. S3, Supporting Material), indicating a good pretreatment and recovery efficiency of the [C2C1Im][OAc]-octanol system.
One of the major challenges in a rapid commercialization of the IL-based lignocellulose pretreatment is the price of ionic liquids. Consequently, an efficient recycle of IL is required. The function of antisolvents in cellulose solvent-based pretreatments is typically for regeneration of dissolved lignocellulose for subsequent enzymatic hydrolysis and fermentation. Most antisolvents are polar and miscible with ionic liquids, making it hard to recycle ionic liquids.
Conclusion
This study demonstrated a new antisolvent process that allows for the treatment of wet biomass, eliminates the use of large quantities of process water, and enables efficient [C2C1Im][OAc] and antisolvent recycle via the use of a multiphase system. The study showed that enzymatic hydrolysis of PSG is independent of antisolvent when alcohols of different alkyl chains are used. However, the polydispersity of recovered lignin is dependent upon the alcohol used as a precipitant, with the octanol system showing the broadest range of lignin components migrating to this antisolvent. The [C2C1Im][OAc]-octanol system enabled high enzymatic glucan digestibilities at a low enzyme dosage and lignin fractionation and, due to its immiscibility with [C2C1Im][OAc] and 10 % water, enabled efficient [C2C1Im][OAc] recycle via this multiphase separation. Solids recovered after enzymatic hydrolysis consisted mainly of lignin which was shown to have undergone depolymerization. Recycled [C2C1Im][OAc] from this system was shown to be an effective pretreatment solvent after at least three reuses, as was the octanol antisolvent, and importantly, it was shown that the resulting PSG samples showed similar chemical and structural characteristics to SG pretreated by fresh [C2C1Im][OAc]. The [C2C1Im][OAc]-octanol process demonstrated in this study therefore shows promise as an efficient new pretreatment system.
References
George A, Tran K, Morgan T, Benke P, Berrueco C, Lorente E, Wu B, Keasling J, Simmons BA, Holmes B (2011) The effect of ionic liquid cation and anion combinations on the macromolecular structure of lignins. Green Chem 13(12):3375–3385
Heikkinen S, Toikka MM, Karhunen PT, Kilpeläinen IA (2003) Quantitative 2D HSQC (Q-HSQC) via suppression of J-dependence of polarization transfer in NMR spectroscopy: application to wood lignin. J Am Chem Soc 125(14):4362–4367
Kim H, Ralph J (2010) Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d6/pyridine-d5. Org Biomol Chem 8(3):576–591
Kim Y, Mosier NS, Ladisch MR, Ramesh Pallapolu V, Lee YY, Garlock R, Balan V, Dale BE, Donohoe BS, Vinzant TB, Elander RT, Falls M, Sierra R, Holtzapple MT, Shi J, Ebrik MA, Redmond T, Yang B, Wyman CE, Warner RE (2011) Comparative study on enzymatic digestibility of switchgrass varieties and harvests processed by leading pretreatment technologies. Bioresour Technol 102(24):11089–11096
Li C, Knierim B, Manisseri C, Arora R, Scheller HV, Auer M, Vogel KP, Simmons BA, Singh S (2010) Comparison of dilute acid and ionic liquid pretreatment of switchgrass: biomass recalcitrance, delignification and enzymatic saccharification. Bioresour Technol 101(13):4900–4906
Mansfield SD, Kim H, Lu F, Ralph J (2012) Whole plant cell wall characterization using solution-state 2D NMR. Nat Protoc 7(9):1579–1589
Mosier N, Wyman C, Dale B, Elander R, Lee Y, Holtzapple M, Ladisch M (2005) Features of promising technologies for pretreatment of lignocellulosic biomass. Bioresour Technol 96(6):673–686
Moxley G, Zhang Y-HP (2007) More accurate determination of acid-labile carbohydrate composition in lignocellulose by modified quantitative saccharification. Energy Fuels 21:3684–3688
Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK (2010) Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3:10
Park S, Johnson D, Ishizawa C, Parilla P, Davis M (2009) Measuring the crystallinity index of cellulose by solid state 13 C nuclear magnetic resonance. Cellulose 16(4):641–647
Rollin JA, Zhu Z, Sathitsuksanoh N, Zhang Y-HP (2011) Increasing cellulose accessibility is more important than removing lignin: a comparison of cellulose solvent-based lignocellulose fractionation and soaking in aqueous ammonia. Biotechnol Bioeng 108(1):22–30
Sathitsuksanoh N, Holtman KM, Yelle DJ, Morgan T, Stavila V, Pelton J, Blanch H, Simmons BA, George A (2014) Lignin fate and characterization during ionic liquid biomass pretreatment for renewable chemicals and fuels production. Green Chem. doi:10.1039/C3GC42295J
Sathitsuksanoh N, Zhu Z, Wi S, Zhang Y-HP (2011) Cellulose solvent based biomass pretreatment breaks highly ordered hydrogen bonds in cellulose fibers of switchgrass. Biotechnol Bioeng 108(3):521–529
Sluiter A, Hames B, Ruiz R, Scarlata C, Sluiter J, Templeton D and Crocker D (2011) Determination of structural carbohydrates and lignin in biomass. Laboratory analytical procedure (LAP). Technical report. NREL/TP-510-42618. http://www.nrel.gov/biomass/pdfs/42618.pdf. Accessed 7 July 2013.
Varanasi P, Singh P, Auer M, Adams PD, Simmons BA, Singh S (2013) Survey of renewable chemicals produced from lignocellulosic biomass during ionic liquid pretreatment. Biotechnol Biofuels 6(1):14
Yelle DJ, Ralph J, Frihart CR (2008) Characterization of nonderivatized plant cell walls using high-resolution solution-state NMR spectroscopy. Magn Reson Chem 46(6):508–517
Zhang Y-HP, Lynd LR (2004) Toward an aggregated understanding of enzymatic hydrolysis of cellulose: noncomplexed cellulase systems. Biotechnol Bioeng 88:797–824
Zhu Z, Sathitsuksanoh N, Vinzant T, Schell DJ, McMillan JD, Zhang Y-HP (2009) Comparative study of corn stover pretreated by dilute acid and cellulose solvent-based lignocellulose fractionation: enzymatic hydrolysis, supramolecular structure, and substrate accessibility. Biotechnol Bioeng 103(4):715–724
Acknowledgments
This work is supported by the Office of Biological and Environmental Research in the DOE Office of Science through the Joint BioEnergy Institute (JBEI) (Contract number DE-AC02-05CH11231). We would like to thank Novozymes® North American for providing CTec2 and HTec2 enzyme mixtures. We were grateful to Professor John Ralph of the Biochemistry Department, University of Wisconsin (USA) for his helpful suggestions on HSQC experiments.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sathitsuksanoh, N., Sawant, M., Truong, Q. et al. How Alkyl Chain Length of Alcohols Affects Lignin Fractionation and Ionic Liquid Recycle During Lignocellulose Pretreatment. Bioenerg. Res. 8, 973–981 (2015). https://doi.org/10.1007/s12155-015-9643-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12155-015-9643-9