
Abstract
Objectives
To retrospectively compare the overall and event-free survival rates of patients with standard and high risk medulloblastoma who received postoperative radiotherapy (RT) followed by maintenance chemotherapy.
Methods
The study included 48 patients with medulloblastoma who were treated and followed-up between 2005 and 2021. Patients were classified according to the Chang classification because no molecular analysis was done. Immediately after surgery all patients received postoperative RT followed by eight cycles of chemotherapy (SIOP/UKCCSG PNET-3 protocol); if thrombocytopenia developed, carboplatin was replaced by cisplatin to avoid treatment delay. The clinical characteristics, risk categories and treatment outcomes of all patients were analyzed.
Results
The mean age of the 48 patients (26 males, 22 females) at diagnosis was 7.27±4.21 y. The median start time of RT after surgery was 37 (range 19–80) d. The median follow-up was 56 (3–216) mo. The 5-year event-free survival was 61.2±10% in the high-risk group and 82.5±11.5% in the standard-risk group. The 5-year overall survival was 73.2±7.1%; it was 61.2±10% and 92.9±6.9% for high- and standard-risk patients, respectively (p = 0.026).
Conclusions
The outcomes of patients who were started on the modified SIOP/UKCCSG PNET-3 chemotherapy protocol, in which RT was begun as soon as possible after surgery, were comparable to those of current treatment protocols. Although a definitive conclusion is difficult, given the limited number of patients in the present study, authors suggest that their treatment protocol is a viable option for centers with limited facilities (such as an inability to perform molecular analysis).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Medulloblastoma (MB) is the most common malignant brain tumor in children [1], and comprises about 20% of all childhood central nervous system tumors. The annual incidence is 5.07 children per million, with bimodal peaks at 3–4 and 7–8 y of age [2]. Patients younger than 3 y of age, with metastasis at diagnosis, or residual tumors >1.5 cm2 in the area are considered high risk [3]. Recently, large cell/anaplastic histopathology and MYC amplification have been identified as high-risk factors, while WNT over-expression has the best prognosis [4]. Here, authors present the clinical characteristics, risk categories, treatments, and outcomes of 48 MB patients who received the SIOP/UKCCSG PNET-3 chemotherapy protocol between 2005 and 2021 in their institution.
Material and Methods
Forty-eight patients diagnosed with MB between 2005 and 2021 who were treated and followed at authors’ institution were reviewed; the last date of data collection was 12 July, 2022. The clinical features, histopathology, treatment modalities, prognostic criteria, and survival rates were analyzed. Primary tumors were evaluated by brain magnetic resonance imaging (MRI). Spinal involvement was explored using spinal MRI preoperatively or 3 wk postoperatively. Cerebrospinal fluid (CSF) was analyzed at least 3 wk postoperatively. The Chang staging system was used: M0 indicates no evidence of gross residual tumor or metastasis, M1 is the presence of microscopic tumor cells in the cerebrospinal fluid, M2 is gross nodular seeding within the central nervous system other than the spinal space, M3 is gross nodular seeding in the spinal subarachnoid space, and M4 is metastasis outside the cerebrospinal axis [5]. The extent of resection and any residual tumor were evaluated by postoperative MRI. Gross/total resection was defined as the absence of a visible tumor on postoperative imaging; subtotal resection was defined as when over 50% of the tumor was removed. The histological subtypes were those of the World Health Organization, but authors were not able to perform molecular classification [6]. Patients with postoperative residual tumors (>1.5 cm2 in area), who were younger than 3 y of age, and metastasis staging ≥M1 (Mrisk) were considered to be at high risk.
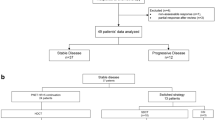
After surgery, all patients older than 3 y of age were prescribed fractionated, intensity modulated external radiotherapy (RT) to both the cranium and spinal cord commencing 3–7 wk after surgery. The craniospinal irradiation (CSI) doses were 23.4 Gy for standard and 36 Gy for high-risk patients and boost doses were delivered to a total of 54 Gy to the primary tumor bed. Radiotherapy was not given to children under 3 y of age; in these children, chemotherapy continued until 3 y of age. If a child received the total chemotherapy protocol dose, temozolomide 200/mg/m2/d was given for 5 d every 4 wk until radiotherapy. Vincristine was administered weekly during RT. Adjuvant chemotherapy was commenced in all patients (both standard and high-risk) approximately 2–3 wk after the end of RT; eight courses of the SIOP/UKCCSG PNET-3 protocol were delivered at an interval of at least 3 wk. In authors’ institution, based on the decision of the multidisciplinary tumor council, the timing of radiotherapy was earlier than in the original protocol; in patients with severe thrombocytopenia, 70 mg/m2 cisplatin was substituted for carboplatin in each course to avoid treatment prolongation (Fig. 1). In patients who relapsed or progressed, “8 drugs in a day” were usually used as salvage chemotherapy and other alternatives.

Patient management diagram
The chemotherapy protocol was conducted over four alternate cycles at a 3-wk interval (eight cycles). The regimen consisted of vincristine 1.5 mg/m2, given three times weekly; etoposide 100 mg/m2/d, given for three consecutive days; and carboplatin 500 mg/m2/d, given on the first two days or vincristine 1.5 mg/m2, given three times weekly; etoposide 100 mg/m2/d, given for three consecutive days; and cyclophosphamide 1.5 g/m2, with mesna given on the first day. Patients who received at least one course of chemotherapy were included in the study, even if they did not complete the treatment.
The study was approved by the Medical Ethics Committee (16/12/2021, no. 2021000540). Informed consent to treat the disease was obtained from the patients’ parents.
IBM SPSS ver. 23 was used for all analyses. Overall survival (OS) was defined as the time from diagnosis to death or the day of the last checkup with the healthcare team. Event-free survival (EFS) was defined as the time from diagnosis to the first recurrence or death. Kaplan–Meier survival curves were drawn and log-rank tests performed to compare OS and EFS based on the known prognostic factors and the timing of RT after surgery. Independent mortality risk factors were analyzed by Cox’s regression. Multivariable analysis was performed for factors with a p value <0.1 in the univariable analyses. The level of significance was taken to be p <0.05.
Results
Twenty-six (54.2%) patients were male and 22 (45.8%) were female. The mean age at diagnosis was 7.27±4.21 (median 6.5, range 1–17) y; 70.8% of the patients (n = 34) underwent their initial surgery at authors’ institution. A second surgery was performed for four patients. At diagnosis, 75% of the patients (n = 36) lacked spinal involvement. Postoperative residues (≥1.5 cm2) were detected in 29.2% of the patients (n = 14). The histology was classical in 72.9% (n = 35) of the patients, desmoplastic nodular in 10.4% (n = 5), and large cell/anaplastic in 16.7% (n = 8). Recurrent disease developed in 18.7% (n = 9), of whom seven died and two are alive. Of the recurrences, five were metastatic and four had local disease (two high risk, two standard risk; one survived and the other three died). The median follow-up was 56 (3–216) mo. All patients under 3 y of age at diagnosis were ultimately given radiotherapy, except one who died before 3 y of age while on chemotherapy.
The 5-year event-free survival of those younger (n = 5) and older (n = 43) than age 3 was 30.0±23.9% and 74.2±7.7%, respectively (P = 0.038). The median start time of radiotherapy after surgery was 37 (range 19–80) d for patients >3 y old. Radiotherapy was interrupted for more than 1 wk in one patient only (now alive). The 5-year event-free survival of patients for delayed (beyond 7 wk) and non-delayed radiotherapy was 59.7±14.6% and 76.7±7.4% (P = 0.385), respectively, without considering the risk group. Table 1 summarizes the clinical characteristics and EFS of the patients.
Overall, 13 of 48 the patients died and 35 are still alive (72.9%); 12 of 30 high-risk patients and 1 of 18 standard-risk patients died. Kaplan–Meier analysis revealed that the overall survival rate was 53.6±16.4%, including 39.0±17.3% for the high-risk and 92.9±6.9% for the standard-risk patients (P = 0.014) (Fig. 2a). Overall 5 y, 73.2±7.1% of the patients survived: 61.2±10% for the high-risk group and 92.9±6.9% for the standard-risk group (P = 0.026) (Fig. 2b). The overall EFS at 5 y was 69.7± 7.6%: 61.2±10% in the high-risk group and 82.5±11.5% in the standard-risk group (P = 0.072).

Overall (a) and 5-year (b) survival of the patients by risk group
Although authors found no significant risk factor effecting survival on univariate Cox regression analysis, the values for residual disease and age risk were borderline (P <0.1). On multivariable analysis, neither residual disease nor age risk were not significant but the value was P <0.1 for residual disease (Table 2).
Three of the 35 surviving patients were followed-up in another hospital; another five were over the age of 21 and had to be followed up at adult centers. The remaining 27 patients were followed regularly. One year after treatment cessation, authors began to check for ototoxicity and detected it in five patients (two at standard risk, three at high risk); it was severe in three and mild in two. One patient had bilateral mild mixed hearing loss and this patient also had Apert syndrome. Both tympanic membranes were perforated in one patient due to chronic otitis, and he had conductive hearing loss severe enough to require a hearing aid; he underwent tympanoplasty. All the patients had neurology referrals, while there was no routine neurocognitive follow-up. The median number of episodes of febrile neutropenia was 3 (0–8) (grade 3/4) and 11 patients did not have any episodes of febrile neutropenia; no one died of febrile neutropenia. Nine patients did not require thrombocyte transfusion and the median transfusion frequency was 3 (0–7) (grade 3/4). No severe bleeding arose due to thrombocytopenia. Table 3 summarizes the adverse effects detected during follow-up.
Discussion
The five medulloblastoma subgroups are defined histopathologically: classical, desmoplastic/nodular, extensive nodular MB, large cell, and anaplastic (the last two were subsequently combined into a single histopathological category) [7]. Medulloblastomas have also been categorized into four molecular subtypes: WNT, SHH, Group 3, and Group 4, according to the clinical and genetic features [8, 9]. As authors could not perform molecular analyses in their patients, they used the Chang Staging System for risk stratification.
It has been shown that the addition of chemotherapy to radiotherapy and completion of RT within 50 d after inception are independent predictors of improved EFS in non-metastatic patients with SIOP/UKCCSG PNET-3 in a multivariable analysis [10]. In a study of HIT-SIOP PNET-4 [11], the median time from diagnosis to RT was 37 d and the prognosis was poorer in patients in whom RT was delayed for over 7 wk. Excellent survival rates were reported in patients with no postoperative residual tumor and no delay in RT. In the present study, authors used SIOP/UKCCSG PNET-3 and initiated radiotherapy a median of 37 d after surgery, together with weekly vincristine. The survival of the present high-risk patients was similar to that reported, while the survival of standard-risk patients was > 80%, consistent with the results of the SIOP PNET-4 study [11]. There were no significant risk factor on multivariable analysis, however, if there were large number of patients, residual disease might be a candidate to be a risk factor because of borderline value (P = 0.081).
In metastatic MB (M2/3) patients who received SIOP/UKCCSG PNET-3 chemotherapy, the OS was 50% at 3 y and 43.9% at 5 y. The EFS was 39.7% at 3 y and 34.7% at 5 y. In this study, the median interval from surgery to RT was 117 (mean 121, range 29–212) d [12]. In the present cases, the mean start of postoperative RT was 37 d after surgery. The 5-year EFS of high-risk patients was 61.2%, while the 5-year EFS of M3 patients with only spinal involvement was 59.5%. These results are better than those of the original chemotherapy protocol, and similar to those of more recent studies in which RT was begun immediately [11, 13].
In developing countries, limited resources, the level of patient awareness, and difficulties in accessing treatment are the main causes of the failure of optimal treatment [14]. To prevent this, the patients in the present study were closely followed; the pediatric oncologists in authors’ institution follow patients on their own personal phones and are in close communication with the patients’ parents to ensure that they come to treatment regularly and undergo optimal management of their complications. When there are difficulties, families can get social assistance from the government. In Table 4, the current treatment outcomes in some centers in developing countries that treat patients without molecular analyses are summarized [15,16,17,18,19,20,21,22,23,24,25,26,27]. Some of these centers mention limited resources regarding treatment access [15, 17, 18, 23, 24]. Some focused on the negative consequences of patients refusing or abandoning treatment [23, 25]. Some centers point out the general problems with malnutrition, infection, and parasitic diseases in developing countries [17, 27], even when all treatment conditions are optimal.
Ototoxicity is an important risk considering the RT doses used in MB protocols; RT and cisplatin may have synergistic toxic effects. Approximately 40–60% of long-term survivors of childhood MB experience moderate to severe hearing loss [28]. In a meta-analysis of 5077 individuals, the prevalence of ototoxic hearing loss associated with carboplatin-only regimens was 13.47% (approximately one-third of that associated with cisplatin alone) [29]. Comparing intensity modulated radiotherapy (IMRT) and standard CSI, IMRT was found to give a reduced dose to the cochlea and grade 3/4 hearing loss occurred in only 13% of the IMRT group (median follow-up 18 mo) compared to 64% of the other group (median follow-up 51 mo) [28]. IMRT treatment has been used in authors’ institution for about 10 y. In present patients, the ototoxicity rate was 18.5% in the patients (5/27) followed up: one patient developed ototoxicity and additional complications after surgery; another had Apert syndrome, which has a deafness component; two had received cisplatin instead of carboplatin due to refractory thrombocytopenia; and one had no risk factors. The lower ototoxicity in present patients might be due to the use of carboplatin and IMRT, but the frequency could increase over time with follow-up.
This study has some limitations. It was a single center study with a small number of patients. The authors could not perform molecular analysis and had to group the patients according to the Chang system. There were no neurocognitive evaluations and it was not a randomized prospective study. The authors have begun to refer their patients for molecular analysis to a feasible center and are considering as part of the risk stratification for the same treatment protocol. As patient survival increases, the long-term effects will become more important, like short stature, infertility, and neurocognitive dysfunction.
In summary, the authors prioritized the RT timing of the original SIOP/UKCCSG PNET-3 protocol and changed carboplatin to cisplatin in patients with thrombocytopenia to eliminate any treatment delay in patients with medulloblastoma. The patients’ outcomes were similar to those of current protocols using molecular studies. Although a definitive conclusion is impossible given the small number of patients, authors suggest that the present protocol is a viable option for centers with limited facilities.
References
McKean-Cowdin R, Razavi P, Barrington-Trimis J, et al. Trends in childhood brain tumor incidence, 1973–2009. J Neurooncol. 2013;115:153–60.
Packer RJ, Macdonald T, Vezina G, Keating R, Santi M. Medulloblastoma and primitive neuroectodermal tumors. Handb Clin Neurol. 2012;105:529–48.
Parkes J, Hendricks M, Ssenyonga P, et al. SIOP PODC adapted treatment recommendations for standard-risk medulloblastoma in low and middle income settings. Pediatr Blood Cancer. 2015;62:553–64.
Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011;29:1400–7.
Dufour C, Beaugrand A, Pizer B, et al. Metastatic medulloblastoma in childhood: Chang’s classification revisited. Int J Surg Oncol. 2012;2012:245385.
Pickles JC, Hawkins C, Pietsch T, Jacques TS. CNS embryonal tumours: WHO 2016 and beyond. Neuropathol Appl Neurobiol. 2018;44:151–62.
Ellison DW. Childhood medulloblastoma: Novel approaches to the classification of a heterogeneous disease. Acta Neuropathol. 2010;120:305–16.
Ramaswamy V, Taylor MD. Medulloblastoma: From myth to molecular. J Clin Oncol. 2017;35:2355–63.
Robinson GW, Rudneva VA, Buchhalter I, et al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): Therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol. 2018;19:768–84.
Taylor RE, Bailey CC, Robinson K, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J Clin Oncol. 2003;21:1581–91.
Lannering B, Rutkowski S, Doz F, et al. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: Results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol. 2012;30:3187–93.
Taylor RE, Bailey CC, Robinson KJ, et al. Outcome for patients with metastatic (M2–3) medulloblastoma treated with SIOP/UKCCSG PNET-3 chemotherapy. Eur J Cancer. 2005;41:727–34.
Parkes J, Hendricks M, Ssenyonga P, et al. SIOP PODC adapted treatment recommendations for standard-risk medulloblastoma in low and middle income settings. Pediatric Blood Cancer. 2015;62:553–64.
Ganguly S, Kinsey S, Bakhshi S. Childhood cancer in India. Cancer Epidemiol. 2021;71:101679.
Ali N, Moustafa A, Emad R, Ebeid E. Treatment outcome and prognostic factors in children with medulloblastoma: A retrospective study of 53 children in a developing country. Egypt Int J Hematol Oncol. 2019;29:22–30.
Sirachainan N, Pakakasama S, Anurathapan U, et al. Outcome of newly diagnosed high risk medulloblastoma treated with carboplatin, vincristine, cyclophosphamide and etoposide. J Clin Neurosci. 2018;56:139–42.
Mehrvar A, Tashvighi M, Hedayati Asl, et al. Management and outcomes of treating pediatric medulloblastoma: An eight years’ experience in an Iranian pediatric center. Childs Nerv Syst. 2018;34:639–47.
Bleil CB, Bizzi JWJ, Bedin A, de Oliveira FH, Antunes ÁCM. Survival and prognostic factors in childhood medulloblastoma: A Brazilian single center experience from 1995 to 2016. Surg Neurol Int. 2019;10:120.
Muzumdar D, Deshpande A, Kumar R, et al. Medulloblastoma in childhood-King Edward Memorial hospital surgical experience and review: Comparative analysis of the case series of 365 patients. J Pediatr Neurosci. 2011;6:S78.
Gupta T, Jalali R, Goswami S, et al. Early clinical outcomes demonstrate preserved cognitive function in children with average-risk medulloblastoma when treated with hyperfractionated radiation therapy. Int J Radiat Oncol Biol Phys. 2012;83:1534–40.
Kumar LP, Deepa SF, Moinca I, Suresh P, Naidu KV. Medulloblastoma: A common pediatric tumor: Prognostic factors and predictors of outcome. Asian J Neurosurg. 2015;10:50.
Gaur S, Kumar SS, Balasubramaniam P. An analysis of medulloblastoma: 10 year experience of a referral institution in South India. Indian J Cancer. 2015;52:575–8.
Wang C, Yuan XJ, Jiang MW, Wang LF. Clinical characteristics and abandonment and outcome of treatment in 67 Chinese children with medulloblastoma. J Neurosurg Pediatr. 2016;17:49–56.
Rajagopal R, Abd-Ghafar S, Ganesan D, et al. Challenges of treating childhood medulloblastoma in a country with limited resources: 20 years of experience at a single tertiary center in Malaysia. J Glob Oncol. 2017;3:143–56.
Das A, Achari RB, Zameer L, Sen S, Krishnan S, Bhattacharyya A. Treatment refusal and abandonment remain major concerns despite good outcomes with multi-modality management in pediatric medulloblastoma: Experience from a cancer center in Eastern India. Indian J Med Paediatr Oncol. 2019;40:S13–5.
Bokun J, Grujicic D, Skender-Gazibara M, et al. Management and treatment of children with medulloblastoma in Serbia, a middle-income country. J Buon. 2018;23:1156–62.
Küpeli S, Sezgin G, Bayram İ, Tanyeli A. Medulloblastoma in children: 84 patients from a single institution. Cukurova Med J. 2020;45:56–62.
Huang E, Teh BS, Strother DR, et al. Intensity-modulated radiation therapy for pediatric medulloblastoma: Early report on the reduction of ototoxicity. Int J Radiat Oncol Biol Phys. 2002;52:599–605.
Dillard LK, Lopez-Perez L, Martinez RX, Fullerton AM, Chadha S, McMahon CM. Global burden of ototoxic hearing loss associated with platinum-based cancer treatment: A systematic review and meta-analysis. Cancer Epidemiol. 2022;79:102203.
Author information
Authors and Affiliations
Contributions
İK, AD, OSD, HKŞ, AU, ŞBG: Constructing the hypothesis or idea of research and/or article, providing personnel, environment, financial support tools that are vital for the study, biological materials, taking responsibility of the referred patients; İK, AD: Planning methodology to reach the conclusions, organizing, supervising the course of progress and taking the responsibility of the research/study, literature review, writing the article, reviewing the article before submission scientifically besides spelling and grammar; İK, AD, OSD, AU, HKŞ: Data collection and/or processing; İK, AD, OSD: Taking responsibility in logical interpretation and conclusion of the results. All authors read and approved the final manuscript. İK will act as guarantor for this manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kartal, İ., Dağdemir, A., Dinçer, O.S. et al. Treatment Outcomes of Childhood Medulloblastoma with the SIOP/UKCCSG PNET-3 Protocol. Indian J Pediatr 90, 1116–1122 (2023). https://doi.org/10.1007/s12098-023-04675-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-023-04675-w