
Abstract
Purpose
The advent of circulating tumor DNA (ctDNA) technology has provided a convenient and noninvasive means to continuously monitor cancer genomic data, facilitating personalized cancer treatment. This study aimed to evaluate the supplementary benefits of plasma ctDNA alongside traditional tissue-based next-generation sequencing (NGS) in identifying targetable mutations and tumor mutational burden (TMB) in colorectal cancers (CRC).
Methods
Our study involved 76 CRC patients, collecting both tissue and plasma samples for NGS. We assessed the concordance of gene mutational status between ctDNA and tissue, focusing on actionable genes such as KRAS, NRAS, PIK3CA, BRAF, and ERBB2. Logistic regression analysis was used to explore variables associated with discordance and positive mutation rates.
Results
In total, 26 cancer-related genes were identified. The most common variants in tumor tissues and plasma samples were in APC (57.9% vs 19.7%), TP53 (55.3% vs 22.4%) and KRAS (47.4% vs 43.4%). Tissue and ctDNA showed an overall concordance of 73.53% in detecting actionable gene mutations. Notably, plasma ctDNA improved detection for certain genes and gene pools. Variables significantly associated with discordance included gender and peritoneal metastases. TMB analysis revealed a higher detection rate in tissues compared to plasma, but combining both increased detection.
Conclusions
Our study highlights the importance of analyzing both tissue and plasma for detecting actionable mutations in CRC, with plasma ctDNA offering added value. Discordance is associated with gender and peritoneal metastases, and TMB analysis can benefit from a combination of tissue and plasma data. This approach provides valuable insights for personalized CRC treatment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Colorectal cancer (CRC) stands as the third most commonly diagnosed cancer and the second leading cause of cancer-related deaths in 2020, accounting for approximately 9.8% (over 1,880,000) of new cancer cases and 9.2% (over 915,000) of cancer fatalities in 2020 [1]. The initiation and progression of CRC are widely attributed to the presence of gene mutations in several oncogenes, prominently including KRAS, NRAS, BRAF, PIK3CA and HER2 (ERBB2), etc. [2,3,4,5,6,7,8,9]. The accurate and dynamic identification of these therapeutically targetable mutations holds significant promise for precision-based personalized treatment in CRC, ultimately enhancing patient outcomes and prognosis.
Tumor tissue next-generation sequencing (NGS) of clinically targetable mutations is important for precise treatment for CRC, while intertumor and intratumor heterogeneity hampers the further application of tissue-based NGS [5, 10]. In addition, tumor tissues are not always available or eligible for NGS. Recently, developed circulating tumor DNA (ctDNA) technique as a convenient and noninvasive means has been rapidly employed for dynamically obtaining and monitoring landscape of genomic information to instruct personalized cancer treatment [11, 12], which has a concordant detection efficacy with the matched tumor tissue NGS [13] but overcomes the influence of intratumor heterogeneity [14] affecting tissue NGS. ctDNA has been effectively applying in the identification of clinically relevant mutations. For example, Hsu et al. targeted ctDNA to monitor genetic variants and response to therapies and predict prognosis in CRC [15, 16]. Tarazona et al. detected plasma post-surgery ctDNA to track minimal residual disease and identify a high risk of relapse in patients with localized colon cancer, which showed post-surgery ctDNA detection was correlated with poor disease- free survival and that presence of ctDNA post therapy in patients receiving adjuvant chemotherapy was associated with early relapse [17]. Xu et al. explored the application of ctDNA in the assessment of clinical tumor mutation burden (TMB) in Chinese patients with metastatic CRC [18].
Not only does ctDNA detect the same mutations as tissue (accounting for most of those detected), but it can also distantly identify some mutations that might be omitted by the other method. For example, Takeda et al. showed that in 34 untreated CRC patients, 53 mutations were detected in tumor tissues, and 47 mutations were detected in ctDNA, 20 of which were undetected in tissues [19]. Cao et al. reported among 59 mutations in 11 advanced CRC tissues, 52 (88.14%) was also identified in matched blood, while 19 mutations in plasma ctDNA were missed in the corresponding tissues [20]. It appears that plasma ctDNA could detect more cancer mutations in CRC than tissues. In addition, detection concordance has been observed between some important clinically relevant gene mutations in tissues and ctDNA. For example, some reports showed a high concordance (77%) of KRAS variant between tumor tissues and plasma [21], and concordance between tissue and blood ctDNA ranged from 63.2% (APC) to 85.5% (BRAF) in CRC [22].
To date, there is a lack of consistency in studies regarding the positive detection of clinically targetable mutations, both at the comprehensive and individual levels, with limited investigations into the application of plasma ctDNA for evaluating TMB status in CRC. In this study, we systematically assess the detection efficacy of tissue versus plasma ctDNA, along with the additional value of plasma ctDNA in comparison to tissue-based NGS, for therapeutically targetable mutations (at both comprehensive and individual levels) and TMB-H in Chinese CRC patients.
Materials and methods
Patients and sample collection
We conducted a retrospective review and enrolled 76 CRC patients who underwent surgical resection at eight hospitals. Peripheral blood samples were collected from these patients prior to treatment, and none had received radio chemotherapy before the sample collection or surgical tumor resection. Additionally, the patients had an adequate quantity and quality of tissue DNA for NGS analysis. Those individuals with concurrent cancer types were excluded from the study. The diagnosis of all the samples was performed by two experienced molecular pathologists based on the morphology of hematoxylin & eosin staining (HE), and the tumor cell content was higher than 50%. This study was approved by the ethics committees of the corresponding hospitals. Written informed consent was obtained from all enrolled patients.
DNA extraction and sequencing
Tissue DNA was extracted from the FFPE tissues using the QIAamp DNA FFPE tissue kit (Qiagen). The plasma DNA was extracted using a DNeasy Blood & Tissue kit (Qiagen) according to the manufacturer’s instructions. The resultant DNA was then quality-controlled using Nanodrop and Qubit (Thermo Fisher Scientific) to ensure adequate purity and quality. Illumina paired-end libraries were prepared from extracted DNA and sequenced on Illumina HiSeq platforms. The 556 or 105 panel produced by Shanghai Tongshu Biotechnology Co., Ltd. was used as a DNA capture probe of cancer-related genes. All the tumor tissues and plasma samples were subjected to NGS of driver mutated genes. The average sequencing depth in tissues is ≥ 1000 × and the average sequencing depth in plasma cfDNA is ≥ 7000 × . The variant allele frequency (VAF) is ≥ 1% for tissue DNA and ≥ 0.1% for cfDNA from plasma. BWA (Burrows-Wheeler-Alignment) software was used to compare the sequencing data. GATK (The Genome Analysis Toolkit), MuTect [23] and VarScan [24] were used to alignment optimization, variant calling and annotation, respectively.
The quantification of ctDNA levels followed a previously established method [25, 26]. This involved calculating the maximum variant allele frequency (maxVAF) and then determining the ctDNA concentration (in haploid genomic equivalents per milliliter, hGE/mL) using the formula: ctDNA concentration (hGE/mL) = (mean ctDNA VAF * cell-free DNA concentration (pg/mL))/3.3, assuming that each haploid genomic equivalent (hGE) weighed 3.3 pg.
Statistical analysis
Detected mutations with allele abundance of ≥ 0.1% were recorded. Samples that were identified with at least one mutation in oncodrivers by any of the tissue and plasma assays were considered true positive, and those showing negative by both assays were considered true negative [27]. The concordance was defined as the number of concordant positive and negative cases/total cases × 100%, positive detection rate was calculated as the positively detected case number/total case number × 100%, and sensitivity was shown as the detected case number of true positive/total true positive case number × 100%. Additionally, the TMB analysis exclusively employed sequencing data from the panel of 556 cancer-related genes, utilizing the upper quartile TMB value of the tissue sample as the threshold to distinguish TMB levels. The software SPSS 25.0 (IBM Corp., Armonk, NY, USA) was used for all statistical analysis. χ2 test was used in the univariate analysis of ctDNA-positive rate and concordance detection. Statistical significance was considered when P < 0.05.
Results
Patient characteristics
Seventy-six CRC patients were successfully enrolled in the study, and tissue and plasma samples were collected and analyzed separately for NGS. Within our cohort, there were 49 males (64.47%) and 27 females (35.53%); ages ranged from 33 to 80 (median: 63). The majority of patients had stage III (48, 63.16%), and the remainder had stage II (11, 14.47%) or IV (17, 22.37%). Fifty-nine patients (77.63%) had no distant metastasis, and 17 patients (22.37%) had distant metastasis at different sites including liver, lung, peritoneum, navel, etc. In addition, 4 patients were DNA mismatch repair (MMR) deficient (dMMR), and 72 were MMR proficient (pMMR), where MMR status was identified by immunohistochemistry (IHC). The basic information of the patients was shown in Table 1.
Concordance of gene mutational status between ctDNA and tissue
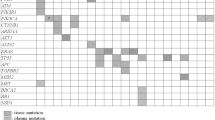
In total, 26 cancer-related genes were found in tissue or plasma samples from these 76 CRC patients. As shown in Fig. 1a, the most frequent variants occurred in APC (57.9% vs 19.7%), TP53 (55.3% vs 22.4%) and KRAS (47.4% vs 43.4%), both in tumor tissue and plasma ctDNA samples. The mutation type of each oncodriver was summarized in Supplementary Table 1. The positive detection rates of tissue and ctDNA were 96.05% and 71.05%, respectively, with an overall consistency of 73.53% (Fig. 1B, C). Here, we focused on the genes KRAS, NRAS, PIK3CA, BRAF and ERBB2, where mutations detected were considered potentially actionable. The positive mutation rate of plasma plus tissue testing for these genes was not lower than that of a single assay, either for individual genes or gene pools as shown in Fig. 2A. Particularly, plasma ctDNA significantly enhances the tissue-based detection of KRAS and KRAS/NRAS/BRAF/PIK3CA/ERBB2 (McNemar’s test, p < 0.01) (Supplementary Table 2). The overall concordance of KRAS, NRAS, PIK3CA, BRAF and ERBB2 between plasma- and tissue-based analyses was 75% (57/76), 90.79% (69/76), 96.05% (73/76), 100% (76/76) and 94.74% (72/76) (Fig. 2b). The concordance analysis of all detected genes is presented in Supplementary Table 3.

Concordance of mutation landscape between tissue and plasma ctDNA. A The mutational landscapes of tissue and plasma ctDNA are provided along with the most frequently mutated oncodrivers. B A comparison of the positive mutation rate of all genes between tissue and plasma ctDNA. C Concordance of all genes between tissue and plasma ctDNA

Concordance of KRAS/PIK3CA/BRAF/NRAS/ERBB2 between tissue and plasma ctDNA. A A comparison of the positive mutation rate of KRAS/PIK3CA/BRAF/NRAS/ERBB2 between tissue and plasma ctDNA. B Concordance of KRAS/PIK3CA/BRAF/NRAS/ERBB2 between tissue and plasma ctDNA. *p < 0.05; **p < 0.01; ns, no significant difference
Variables associated with discordance and positive mutation rate
In addition, variables associated with the discordance and positive mutation rate of tissue and ctDNA were analyzed. The overall positive mutation rate of KRAS/PIK3CA/BRAF/NRAS/ERBB2 for advanced patients was higher than early patients, and those with distal metastasis were higher than those without distal metastasis, but there was no statistically significant difference (Fig. 3). The increased positive mutation rate of combined tissue and plasma testing was independent of the clinical characteristics of the patients (Table 2). We investigated the logistic regression analysis for identifying the variables associated with the discordance. Stage and metastatic status did not appear to be significantly associated with inconsistency (Fig. 3 and Table 3). The discordance showed a strong association with gender (P = 0.030) and peritoneal metastases (P = 0.045).

Variables associated with discordance and positive mutation rate of KRAS/PIK3CA/BRAF/NRAS/ERBB2 between tissue and plasma ctDNA. A The positive mutation rate of KRAS/PIK3CA/BRAF/NRAS/ERBB2 between tissue and plasma ctDNA from patients with different stages. B Concordance of KRAS/PIK3CA/BRAF/NRAS/ERBB2 between tissue and plasma ctDNA from patients with different stages. C The positive mutation rate of KRAS/PIK3CA/BRAF/NRAS/ERBB2 between tissue and plasma ctDNA from patients with distal metastasis or not. D Concordance of KRAS/PIK3CA/BRAF/NRAS/ERBB2 between tissue and plasma ctDNA from patients with distal metastasis or not
Concordance of TMB-H between ctDNA and tissue
TMB was then classified into TMB-H and TMB-L according to the upper quartile TMB of 9.48 mutations/Mb (TMB ≥ 9.48 was defined as TMB-H and those < 9.48 as TMB-L). For the detection of TMB-H, the positive detection rates of TMB-H by tissues and plasma were 25% (16/64) and 7.81% (5/64), respectively, and plasma plus tissue increased the detection rate to 32.81% ([16 + 5]/64), and the overall concordance was 67.19% (43/64) (Fig. 4 and Supplementary Table 4).

Concordance of TMB-H between tissue and plasma ctDNA. A Comparison of the positive detection rate of TMB-H between tissue and plasma ctDNA. B Concordance of TMB status between tissue and plasma ctDNA
4 Discussion
Currently, studies regarding the positive detection of clinically targetable mutations in CRC by ctDNA, at both the whole and individual mutation levels, are not consistent, and there were few studies reporting the application of plasma ctDNA in monitoring TMB in CRC. In this study, we aimed to assess the concordance between gene mutational status in ctDNA and tissue samples from CRC patients and to determine the impact of combining these two modalities in detecting actionable mutations. Our findings shed light on the utility of plasma ctDNA as a complementary tool for assessing genetic alterations in CRC patients. We also evaluated the concordance of TMB-H classification between ctDNA and tissue samples. The progression of driver gene alterations in CRC represents a stepwise tumorigenesis process. It’s noteworthy that less than 1% of human genes are likely to undergo transformation into cancer-driver genes, which play an active role in regulating cell survival and fate, consequently impacting the stability of normal genomes [28, 29]. In contrast to diseases like cystic fibrosis or muscular dystrophy, where cancer does not stem from a single gene defect, it’s more accurate to regard altered cancer genes as contributory factors rather than root causes of cancer. Nevertheless, studies have revealed that several frequently mutated genes in CRC, such as APC, TP53, KRAS, and BRAF, are not only significantly influenced by individual somatic mutations but also exert a substantial functional impact [30]. In our investigation, we identified a total of 26 cancer-related genes, with APC, TP53, and KRAS exhibiting the most prevalent mutations in both tumor tissue and plasma samples. The APC gene is recognized as the sentinel gene in CRC [31]. KRAS plays a pivotal role in promoting cancer through the activation of RAF-MAPK and PI3K pathways. Typically, APC mutations coincide with KRAS or TP53 mutations, or both, as corroborated by our findings. The overall concordance in detecting mutations in these tumor-related genes stood at 73.53%, signifying robust consistency between tissue- and plasma-based NGS. This level of concordance surpasses that reported in some earlier studies [18, 32]. Additionally, we identified other potentially actionable target genes, including NRAS, PIK3CA, BRAF, and ERBB2, which are closely linked to anti-EGFR resistance. The concordance between individual gene mutations from tissue- and plasma-based NGS exceeded 90%, with NRAS achieving a perfect 100% agreement, slightly exceeding results in certain prior studies [33,34,35]. Furthermore, even though the positive mutation rates of KRAS, BRAF, PIK3CA, and ERBB2 were higher in tissue samples, the combination of plasma and tissue data resulted in a higher overall positive detection rate. This underscores that both tissue and plasma ctDNA NGS are effective in identifying therapeutically targetable mutations in CRC. Notably, the increased positive mutation rate achieved through the combined tissue and plasma testing was independent of the clinical characteristics of the patients. In summary, plasma ctDNA adds significant value to routine tissue NGS, as illustrated in Fig. 5.

Schematic diagram of the study
We conducted a comprehensive analysis of variables influencing concordance. Discordance could potentially arise from the low levels of ctDNA shedding by tumors. Our investigation revealed a higher positive mutation rate of KRAS/PIK3CA/BRAF/NRAS/ERBB2 in advanced-stage patients compared to early-stage patients. Likewise, patients with distal metastasis exhibited a higher positive mutation rate than those without distal metastasis, although this difference did not reach statistical significance. This observation suggests that later disease stages may yield more ctDNA release than earlier stages. Notably, the logistic regression analysis revealed that peritoneal metastases were a significant variable associated with discordance, while liver and lung metastases showed no significant association with discordance in our cohort. Hideaki Bando et al. found that lung metastasis alone was the most significant factor associated with discordance [34]. Due to our limited sample size, the difference in discordance between patients with lung metastasis and patients with peritoneal metastasis was only one person. Therefore, these findings should be further substantiated with a larger cohort in future studies. Additionally, the plausible reason for discordance could be lower ctDNA shedding from different tumors [34]. Some studies aslo reported that in recurrence CRCs, ctDNA detection was challenging for lung metastases and peritoneal metastases [34, 36]. This discordance between tissue- and plasma-based NGS is not the factors mentioned above; it’s also influenced by the patients’ treatment history or the differences in tumor heterogeneity, even the bias in multicenter patients [34,35,36].
To account for spatial and temporal variability, it is imperative to periodically assess the genomic profile of CRC patients throughout their treatment. Liquid biopsies can play a pivotal role in profiling the patient’s specific molecular makeup, particularly when considering anti-EGFR treatment options [37, 38]. One of the strongest arguments favoring this minimally invasive approach is the ability to perform RAS/RAF testing at the point of decision-making. Within our cohort, we observed instances where 11 and 2 patients had tissue mutations in KRAS and BRAF that were undetectable in plasma, and conversely, eight and one patients had plasma mutations in KRAS and BRAF that were absent in tissue samples. The absence of RAS/RAF mutations in plasma might be attributed to biological factors influencing ctDNA release—a crucial area for further investigation. This issue becomes even more pronounced in light of false-negative results, which pose a significant challenge in plasma testing for RAS/RAF mutations. Negative interactions may occur between anti-EGFR drugs and oxaliplatin-based regimens in patients with RAS/RAF mutations. Similar trends were observed in the detection of other genes, including PIK3CA and ERBB2 mutations, within our cohort. Additionally, the detection rate of TMB-H in tissue samples was 25%, significantly lower at 7.81% in plasma, with poor consistency. We suspect several factors may contribute to this result. In our study, a significant proportion of patients (59 cases, 77.6%) were classified as stage II/III. The observed low TMB values in ctDNA may be attributed to restrictions on the release of tumor-derived DNA into the bloodstream and factors related to the early stages of the disease. Consequently, the TMB levels in ctDNA (with 92.2% of patients in this study having TMB < 10 mutations/Mb) are significantly lower than tissue TMB levels. In the absence of uniform TMB grading criteria, it is necessary to determine the cutoff value for defining TMB as "TMB-high," which may also be one of the reasons for the bias. Furthermore, compared to TMB estimates based on tumor tissue, TMB estimates based on ctDNA cannot effectively avoid interference from lineage mutations; thus, it may lead to calculation biases, especially in patients with relatively low mutation counts. Despite this, the combined analysis of plasma and tissue increased the positive detection rate to 32.81%. This outcome underscores that for detecting TMB-H in CRC, plasma ctDNA alone may not suffice; nevertheless, it effectively enhances the positive detection rate when used in conjunction with tissue samples. Studies have established a correlation between TMB and ctDNA levels. Lower TMB values are associated with a higher likelihood of false-negative plasma ctDNA results and reduced concordance between tissue and plasma ctDNA detection [39].
Conclusion
Our study highlights the potential clinical implications of combining tissue and plasma-based genetic testing in CRC patients. The high concordance of actionable gene mutations and the increased detection of high TMB suggest that this approach may guide treatment decisions more effectively. The integration of ctDNA analysis in clinical practice may improve the precision and efficacy of treatment strategies for CRC patients.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author upon reasonable request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. https://doi.org/10.3322/caac.21660.
Cremolini C, Rossini D, Dell’Aquila E, Lonardi S, Conca E, Del Re M, et al. Rechallenge for patients with RAS and BRAF wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: a phase 2 single-arm clinical trial. JAMA Oncol. 2019;5(3):343–50. https://doi.org/10.1001/jamaoncol.2018.5080.
Chu YC, Tsai TY, Yadav VK, Deng L, Huang CC, Tzeng YM, et al. 4-Acetyl-antroquinonol B improves the sensitization of cetuximab on both kras mutant and wild type colorectal cancer by modulating the expression of Ras/Raf/miR-193a-3p signaling axis. Int J Mol Sci. 2021;22(14):7508. https://doi.org/10.3390/ijms22147508.
Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533–52. https://doi.org/10.1038/s41573-020-0068-6.
Punt CJ, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. 2017;14(4):235–46. https://doi.org/10.1038/nrclinonc.2016.171.
Uchi R, Takahashi Y, Niida A, Shimamura T, Hirata H, Sugimachi K, et al. Integrated multiregional analysis proposing a new model of colorectal cancer evolution. PLoS Genet. 2016;12(2):e1005778. https://doi.org/10.1371/journal.pgen.1005778.
Algars A, Sundstrom J, Lintunen M, Jokilehto T, Kytola S, Kaare M, et al. EGFR gene copy number predicts response to anti-EGFR treatment in RAS wild type and RAS/BRAF/PIK3CA wild type metastatic colorectal cancer. Int J Cancer. 2017;140(4):922–9. https://doi.org/10.1002/ijc.30507.
Nam SK, Yun S, Koh J, Kwak Y, Seo AN, Park KU, et al. BRAF, PIK3CA, and HER2 oncogenic alterations according to KRAS mutation status in advanced colorectal cancers with distant metastasis. PLoS ONE. 2016;11(3): e0151865. https://doi.org/10.1371/journal.pone.0151865.
La Salvia A, Lopez-Gomez V, Garcia-Carbonero R. HER2-targeted therapy: an emerging strategy in advanced colorectal cancer. Expert Opin Investig Drugs. 2019;28(1):29–38. https://doi.org/10.1080/13543784.2019.1555583.
Saito T, Niida A, Uchi R, Hirata H, Komatsu H, Sakimura S, et al. A temporal shift of the evolutionary principle shaping intratumor heterogeneity in colorectal cancer. Nat Commun. 2018;9(1):2884. https://doi.org/10.1038/s41467-018-05226-0.
Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223–38. https://doi.org/10.1038/nrc.2017.7.
Zill OA, Banks KC, Fairclough SR, Mortimer SA, Vowles JV, Mokhtari R, et al. The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res. 2018;24(15):3528–38. https://doi.org/10.1158/1078-0432.Ccr-17-3837.
Rothwell DG, Ayub M, Cook N, Thistlethwaite F, Carter L, Dean E, et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat Med. 2019;25(5):738–43. https://doi.org/10.1038/s41591-019-0380-z.
Lebofsky R, Decraene C, Bernard V, Kamal M, Blin A, Leroy Q, et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol Oncol. 2015;9(4):783–90. https://doi.org/10.1016/j.molonc.2014.12.003.
Hsu HC, Lapke N, Wang CW, Lin PY, You JF, Yeh CY, et al. Targeted sequencing of circulating tumor DNA to monitor genetic variants and therapeutic response in metastatic colorectal cancer. Mol Cancer Ther. 2018;17(10):2238–47. https://doi.org/10.1158/1535-7163.MCT-17-1306.
Salvianti F, Gelmini S, Mancini I, Pazzagli M, Pillozzi S, Giommoni E, et al. Circulating tumour cells and cell-free DNA as a prognostic factor in metastatic colorectal cancer: the OMITERC prospective study. Br J Cancer. 2021;125(1):94–100. https://doi.org/10.1038/s41416-021-01399-6.
Tarazona N, Gimeno-Valiente F, Gambardella V, Zuñiga S, Rentero-Garrido P, Huerta M, et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann Oncol. 2019;30(11):1804–12. https://doi.org/10.1093/annonc/mdz390.
Xu X, Yu Y, Shen M, Liu M, Wu S, Liang L, et al. Role of circulating free DNA in evaluating clinical tumor burden and predicting survival in Chinese metastatic colorectal cancer patients. BMC Cancer. 2020;20(1):1006. https://doi.org/10.1186/s12885-020-07516-7.
Takeda K, Yamada T, Takahashi G, Iwai T, Ueda K, Kuriyama S, et al. Analysis of colorectal cancer-related mutations by liquid biopsy: utility of circulating cell-free DNA and circulating tumor cells. Cancer Sci. 2019;110(11):3497–509. https://doi.org/10.1111/cas.14186.
Cao W, Xu Y, Chang L, Gong Y, Li L, Mo X, et al. Genotyping of circulating tumor DNA reveals the clinically actionable mutation landscape of advanced colorectal cancer. Mol Cancer Ther. 2019;18(6):1158–67. https://doi.org/10.1158/1535-7163.Mct-18-1247.
Rodriguez-Casanova A, Bao-Caamano A, Lago-Leston RM, Brozos-Vazquez E, Costa-Fraga N, Ferreiros-Vidal I, et al. Evaluation of a targeted next-generation sequencing panel for the non-invasive detection of variants in circulating DNA of colorectal cancer. J Clin Med. 2021;10(19):4487. https://doi.org/10.3390/jcm10194487.
Kato S, Schwaederlé MC, Fanta PT, Okamura R, Leichman L, Lippman SM, et al. Genomic assessment of blood-derived circulating tumor DNA in patients with colorectal cancers: correlation with tissue sequencing, therapeutic response, and survival. JCO Precis Oncol. 2019;3:PO.18.00158. https://doi.org/10.1200/po.18.00158.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. https://doi.org/10.1101/gr.107524.110.
Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25(17):2283–5. https://doi.org/10.1093/bioinformatics/btp373.
Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017;7(12):1394–403. https://doi.org/10.1158/2159-8290.CD-17-0716.
Mao X, Zhang Z, Zheng X, Xie F, Duan F, Jiang L, et al. Capture-based targeted ultradeep sequencing in paired tissue and plasma samples demonstrates differential subclonal ctDNA-releasing capability in advanced lung cancer. J Thorac Oncol. 2017;12(4):663–72. https://doi.org/10.1016/j.jtho.2016.11.2235.
Lin LH, Allison DHR, Feng Y, Jour G, Park K, Zhou F, et al. Comparison of solid tissue sequencing and liquid biopsy accuracy in identification of clinically relevant gene mutations and rearrangements in lung adenocarcinomas. Mod Pathol. 2021;34(12):2168–74. https://doi.org/10.1038/s41379-021-00880-0.
Garraway Levi A, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. https://doi.org/10.1016/j.cell.2013.03.002.
Raskov H, Søby JH, Troelsen J, Bojesen RD, Gögenur I. Driver gene mutations and epigenetics in colorectal cancer. Ann Surg. 2020;271(1):75–85. https://doi.org/10.1097/sla.0000000000003393.
Moshawih S, Lim AF, Ardianto C, Goh KW, Kifli N, Goh HP, et al. Target-based small molecule drug discovery for colorectal cancer: a review of molecular pathways and in silico studies. Biomolecules. 2022;12(7):878. https://doi.org/10.3390/biom12070878.
Joseph R, Little P, Hayes DN, Lee MS. Characterization of the number and site of APC mutations in sporadic colorectal cancer. Alexandria: American Society of Clinical Oncology; 2017.
Cao H, Liu X, Chen Y, Yang P, Huang T, Song L, et al. Circulating tumor DNA is capable of monitoring the therapeutic response and resistance in advanced colorectal cancer patients undergoing combined target and chemotherapy. Front Oncol. 2020;10:466. https://doi.org/10.3389/fonc.2020.00466.
Grasselli J, Elez E, Caratu G, Matito J, Santos C, Macarulla T, et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann Oncol. 2017;28(6):1294–301. https://doi.org/10.1093/annonc/mdx112.
Bando H, Kagawa Y, Kato T, Akagi K, Denda T, Nishina T, et al. A multicentre, prospective study of plasma circulating tumour DNA test for detecting RAS mutation in patients with metastatic colorectal cancer. Br J Cancer. 2019;120(10):982–6. https://doi.org/10.1038/s41416-019-0457-y.
Jiang HQ, Wang BL, Guo W. Analysis of influencing factors of KRAS/NRAS/BRAF/PIK3CA gene mutation consistency in patients with advanced colorectal cancer. Zhonghua Yi Xue Za Zhi. 2021;101(6):400–4. https://doi.org/10.3760/cma.j.cn112137-20200721-02176.
Vidal J, Muinelo L, Dalmases A, Jones F, Edelstein D, Iglesias M, et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann Oncol. 2017;28(6):1325–32. https://doi.org/10.1093/annonc/mdx125.
Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21(7):827. https://doi.org/10.1038/nm0715-827b.
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–6. https://doi.org/10.1038/nature11156.
Bhangu JS, Beer A, Mittlbock M, Tamandl D, Pulverer W, Schonthaler S, et al. Circulating free methylated tumor DNA markers for sensitive assessment of tumor burden and early response monitoring in patients receiving systemic chemotherapy for colorectal cancer liver metastasis. Ann Surg. 2018;268(5):894–902. https://doi.org/10.1097/SLA.0000000000002901.
Acknowledgements
This study was supported by the Xiamen Municipal Bureau of Science and Technology (No: 3502Z20214ZD1018 and 3502Z20209153) and the Medical Innovation Project of Fujian Provincial Health Commission (2021CXB019). The authors thank Shanghai Tongshu Biotechnology Co., Ltd. for technical support.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. All authors participated in manuscript writing and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
This study was approved by the ethics committees of all the participating hospitals.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
All the authors agree to the publication clause.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, W., Huang, Y., Kong, J. et al. Plasma ctDNA enhances the tissue-based detection of oncodriver mutations in colorectal cancer. Clin Transl Oncol 26, 1976–1987 (2024). https://doi.org/10.1007/s12094-024-03422-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-024-03422-7