
Abstract
The acronym for the CCN family was recently revised to represent “cellular communication network”. These six, small, cysteine-enriched and evolutionarily conserved proteins are secreted matricellular proteins, that convey and modulate intercellular communication by interacting with structural proteins, signalling factors and cell surface receptors. Their role in the development and physiology of musculoskeletal system, constituted by connective tissues where cells are interspersed in the cellular matrix, has been broadly studied. Previous research has highlighted a crucial balance of CCN proteins in mesenchymal stem cell commitment and a pivotal role for CCN1, CCN2 and their alter ego CCN3 in chondrogenesis and osteogenesis; CCN4 plays a minor role and the role of CCN5 and CCN6 is still unclear. CCN proteins also participate in osteoclastogenesis and myogenesis. In adult life, CCN proteins serve as mechanosensory proteins in the musculoskeletal system providing a steady response to environmental stimuli and participating in fracture healing. Substantial evidence also supports the involvement of CCN proteins in inflammatory pathologies, such as osteoarthritis and rheumatoid arthritis, as well as in cancers affecting the musculoskeletal system and bone metastasis. These matricellular proteins indeed show involvement in inflammation and cancer, thus representing intriguing therapeutic targets. This review discusses the current understanding of CCN proteins in the musculoskeletal system as well as the controversies and challenges associated with their multiple and complex roles, and it aims to link the dispersed knowledge in an effort to stimulate and guide readers to an area that the writers consider to have significant impact and relevant potentialities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
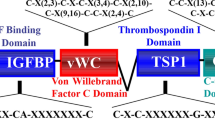
CCN proteins were first discovered in the late 1980s and early 1990s as small proteins (30–40 kDa) enriched in cysteine content (10% by mass, 34–38 conserved residues) and characterized by a tetramodular structure. The first reports described them as growth factors due to their ability to control adhesion, proliferation, migration, apoptosis, and extracellular matrix deposition in diverse cell types (for a review see (Perbal 2004)). As research proceeded, CCN members were defined as matricellular proteins, highlighting their secretory nature and their role in conveying/modulating paracrine stimuli by residing in the extracellular matrix (ECM) and interacting with structural proteins, proper signalling factors and cell surface receptors (Rachfal and Brigstock 2005). More recently, the acronym CCN was revised from the historical initials of the first three family members (Cyr61 = CCN1, CTGF = CCN2 and Nov = CCN3) to the novel concept of the cellular communication network, again underscoring the central role of CCN family members in intercellular communication and fine-tuning of the cellular phenotype thanks to their tightly coordinated pattern of expression (Perbal 2018). Cartilage, bone, and skeletal muscles are connective tissues where cells are interspersed in the ECM; in such tissues, which also need to adapt to constant mechanical stimuli, cellular communication mediated by matricellular proteins if of obvious and paramount importance. Indeed, involvement of CCN family members in the physiology of the musculoskeletal system was already identified in the very first reports (Ivkovic et al. 2003; O’Brien and Lau 1992; Wong et al. 1997). Degenerative pathologies of the musculoskeletal system, such as osteoarthritis and rheumatoid arthritis represent a heavy burden on patients’ quality of life and have high social health costs (Martel-Pelletier et al. 2016; Smolen et al. 2018); data on the emerging role of CCN proteins in their pathogenesis may offer therapeutic and regenerative options. Bone sarcomas of the musculoskeletal system, though representing less than 1% of diagnosed tumours per year (Siegel et al. 2021), are highly aggressive cancers that have a tendency for widespread metastasis and often affect children and young adults (Reed et al. 2017). Bone is the preferential site of metastasis of the most common carcinomas in both sexes and bone metastases often lead to skeletal morbidity, such as pathological fracture, pain, spinal cord compression and hypercalcaemia. Skeletal metastasis is associated with reduced patient quality of life, elevated medical costs and ultimately reduced overall survival (Coleman et al. 2020). Matricellular proteins, as CCN family members, have been demonstrated to be involved in every hallmark of cancer (Chong et al. 2012; Yeger and Perbal 2021). The present review aims to provide a comprehensive view of CCN proteins in the development, physiology, pathology, and malignancy of the musculoskeletal system.
CCN proteins during intrauterine development and differentiation
CCN gene family members are conserved among vertebrates in term of amino acid sequences, protein structures and genetic loci. Bioinformatic analysis revealed an expansion from the sole homologue found in amphioxus, cestode, oyster, and fruit fly to the 4 homologs in lamprey, 5 in ascidian and 9 in zebrafish. Six members belong to this family in mammals, such as mice and humans (Hu et al. 2019) (for a review see Katsube et al. 2009). Given the conservation of the CCN family over evolution, a crucial role of these proteins in vertebrate development can be predicted and has indeed been demonstrated thanks to transgenic mice (Fig. 1) and a description of the expression pattern in the developing musculoskeletal system of vertebrate embryos (Table 1). Only the first two members of the family, CCN1 and CCN2 have been shown to be essential for global embryo development (Ivkovic et al. 2003; Lambi et al. 2012; Mo et al. 2002) and only the expression of CCN2 and CCN3 proved to be essential for correct skeletal development (Heath et al. 2008; Ivkovic et al. 2003; Lambi et al. 2012) (Fig. 1). Therefore, an ancillary role for CCN4, CCN5 and CCN6 in skeletogenesis can be speculated and indeed corresponding knockout mice were viable, fertile, and displayed mild or no skeletal abnormalities (Jiang et al. 2018; Kutz et al. 2005; Maeda et al. 2015). Except for CCN6 (Witte et al. 2009), the expression of all family members has been detected in murine and/or human embryos; CCN2 is expressed solely in active chondrocytes (Friedrichsen et al. 2003; Ivkovic et al. 2003; Kireeva et al. 1997), CCN4 is expressed only in bony elements (French et al. 2004; Witte et al. 2009), and CCN1, CCN3, CCN5 are found in both (Jones et al. 2007; Kanyama et al. 2013; Kireeva et al. 1997; Mo et al. 2002; O’Brien and Lau 1992). Embryonal skeletal muscles expressed low levels of CCN2, CCN3 and CCN5 (Jones et al. 2007; Kocialkowski et al. 2001).

General and skeletal phenotype of mice knockout for different CCN proteins
Undifferentiated bone-marrow stem cells express all CCN family members at the mRNA level (Djouad et al. 2007; Schutze et al. 2005). Embryonic and residual adult mesenchymal stem cells (MSCs) are pluripotent precursors defined by the ability to differentiate along adipocytic, chondrocytic and osteocytic lineages. Commitment and cell fate decisions arise from tightly regulated intra- and extracellular signals from both adjacent cells and niche microenvironment. It is therefore not surprising that matricellular CCN proteins under the guise of cellular communication networks are tightly and coordinately modulated during differentiation of MSCs. CCN1 expression decreases during osteogenic differentiation, whereas the expression of the other family members remains constant. During adipogenic commitment CCN1 and CCN5 expression is progressively lost, whereas CCN2 expression slightly decreases (Schutze et al. 2005). Chondrogenic differentiation results from loss of CCN1 expression, decreased CCN2 and CCN6 expression, and upregulation of CCN3 and CCN4 expression (Djouad et al. 2007; Schutze et al. 2005). Many very recent works have also shed light on the pivotal role of CCN2 in tendogenesis. CCN2 promotes clonal expansion, migration and differentiation of the tendon-derived stem cell population synergizing with BMP12 and TGFβ and reversing the detrimental effects of IL1β on collagen deposition (Lee et al. 2015; Liu et al. 2015a, b; Tarafder et al. 2017). CCN2 decreases with age in rat tendon-derived progenitor cells and its exogenous administration rescues these cells from senescence (Lee et al. 2015; Liu et al. 2015a, b; Rui et al. 2019; Tarafder et al. 2017).
Experimental imbalances of CCN protein levels can influence MSC viability. Indeed, exogenous administration of CCN2 decreases the expression of stemness markers (Lee et al. 2010a, b) and CCN4 is required for murine bone marrow-MSC viability as it mediates BMP-3-induced MSC proliferation (Cernea et al. 2016; Schlegelmilch et al. 2014). CCN4 also constitutes a barrier for mouse embryonic cell reprogramming, which requires downregulation of its expression together with that of byglican via upregulation of miR-135b expression (Li et al. 2014). CCN proteins can skew cellular commitment, as well. MSCs derived from CCN1OCN mice, with targeted disruption of CCN1 under the osteocalcin promoter, display reduced osteogenic potential in favour of adipogenic differentiation in vitro (Zhao et al. 2018). Exogenous administration of CCN2 hinders osteogenic and chondrogenic commitment in favour of fibroblastic commitment in human MSCs (Lee et al. 2010a) and skews adipose stem cell toward tendogenic differentiation sustaining proliferation and increasing tendon markers detrimental to expression of osteogenic marker Runx2 (Li et al. 2019). High levels of CCN5 hinder adipogenic differentiation as they dedifferentiate 3T3-L1 adipose cells into pluripotent myofibroblasts expressing α-SMA (Grünberg et al. 2014).
CCN proteins in the physiology of musculoskeletal system
The vertebrate skeleton is formed through the alternative processes of endochondral and intramembranous ossification (for a review see (Salhotra et al. 2020)). The latter consists of direct differentiation of osteoblasts from MSCs. Endochondral ossification requires the formation of a cartilaginous anlage; during chondrogenesis resting chondroprogenitor cells initially aggregate together to form nodules, proliferate, deposit matrix and terminally differentiate into hypertrophic chondrocytes depositing a calcified matrix. Cartilaginous anlage is later eroded by osteoclasts, multinucleated cells differentiating from macrophages during the process of osteoclastogenesis (for a review see (Xu and Teitelbaum 2013)), allowing invasion of capillaries that convey osteoblast progenitors. Eventually, erosion of cartilaginous anlage and substitution with bone are completed. Endochondral ossification is also a process involved in bone repair. Skeletal muscle cells are also derived from mesenchymal precursors direct differentiation, which requires the proliferation and migration of myoblasts, expressing MyoD and Myf5, subsequent differentiation and expression of specific myogenic markers such as myogenin and MRFs and eventually fusion into myotubes expressing MHC and α-SMA (for a review see (Chal and Pourquié 2017)).
After development, CCN proteins are responsible for maintaining tissue homeostasis in the musculoskeletal system, which undergoes mechanical load and is prone to damage throughout life. These proteins constitute a mechanosensory family, which allows cells to adapt to ever changing conditions modulating the characteristics of the ECM; experimental protocols and cells subjected to mechanical load are extremely varied, indicating that the enhanced expression of CCN proteins upon mechanical stress is likely conserved in the different cytotypes of musculoskeletal system. Notably, in most experimental settings, upregulation of CCN proteins is detected a few hours after mechanical load providing a steady response to changing environment.
Chondrogenesis
Expression of all CCN family members was induced during experimental chondrogenesis in vitro, with peaks at day 28 or beyond, except for CCN3, which was transiently expressed during only the initial phases of chondrogenesis (Kawaki et al. 2008). Most studies reported below were performed in HCS-2/8 cells, a chondrosarcoma-derived cell line that retains the ability to undergo chondrogenic differentiation and confirmed in primary chondrocytes of both human and animal origin.
CCN1 and CCN2 induce chondrogenesis and promote cell adhesion, cellular aggregation, and node formation, inducing aggrecan and type II collagen with consequent increased proteoglycan synthesis and cartilaginous matrix deposition (Hoshijima et al. 2006; Nishida et al. 2007; Sumiyoshi et al. 2013; Wong et al. 1997) (Fig. 2). A pivotal role of CCN2 in chondrocytes has been proven as this matricellular protein also induces proliferation, DNA synthesis and expression of Sox-9, controls glycolysis and metabolic processes (Maeda-Uematsu et al. 2014; Murase et al. 2016), inhibits ALP activity and protects chondrocytes from apoptosis and autophagy (Fujisawa et al. 2008; Hall-Glenn et al. 2013; Nakanishi et al. 2000; Nakao et al. 2005; Nishida et al. 2002; Xing et al. 2018; Yosimichi et al. 2001). CCN2 is regulated in concert with other CCN family members (Kawaki et al. 2008; Maeda-Uematsu et al. 2014); different growth factors and membrane receptors such as BMP2 and TGFβ (Nakanishi et al. 1997; Parada et al. 2013; Song et al. 2007), M-CSF (Nakao et al. 2005), the actin remodelling pathway (Woods et al. 2009) and FGF1 (El-Seoudi et al. 2017); mir-26a and mir-18a (Etich et al. 2015; Ohgawara et al. 2009); and hypoxic microenvironmental conditions (Nishida et al. 2009). The reader is referred to (Nishida and Kubota 2020) for a more detailed overview of CCN2 role in chondrogenesis. Consistent with its anabolic role in chondrocytes, CCN2 is up regulated in response to mechanical stimuli (Furumatsu et al. 2012, 2013; Nishida et al. 2008). Notably, CCN2 expression is decreased in young chondrocytes exposed to an excessive bout of force (> 15 kPa, 30 cycles/min) and in aged bovine chondrocytes, which consistently show decreased ability to respond to mechanical stimuli (Madej et al. 2016). The anabolic role of CCN2 in chondrocytes can also be exploited for regenerative purposes (Nishida et al. 2004).

Schematic representation of the role of different CCN proteins in chondrogenesis
CCN4 appears to have a role similar to that of CCN1 and CCN2 in chondrogenesis; its overexpression in MSCs induces chondrogenesis by binding TGFβ3 and exerting synergistic effect during the activation of Smad2/3 pathways leading to enhanced expression of Sox-9, collagen type II and aggrecan (Fig. 2). Consistent with a pro-chondrogenic role, CCN4-/- mouse cartilage displays reduced histological levels of Sox-9, collagen type II and aggrecan as well as reduced repair after full thickness damage (Yoshioka et al. 2016). According to another report, CCN4 synergizes with Wnt3a in inhibiting TGFβ-induced Smad2/3 pathways in P2 cells and human G6 chondrocytes and skewing cellular phenotype towards osteogenesis (van den Bosch et al. 2014). The alternative role of CCN4 may depend on the upstream activating pathway as well as expression of splicing variants detected in HCS-2/8 cells (Yanagita et al. 2007).
CCN3 counteracts the proliferative potential of CCN2 as it keeps terminally differentiated chondrocytes in a quiescent state and its exogenous administration, overexpression or in vivo supplementation inhibits chondrocyte proliferation and proteoglycan deposition and induces type II collagen, type X collagen, tenascin, lubricin and TGFβ2 expression, whereas its silencing yields the opposite results (Janune et al. 2011b, 2017; Kawaki et al. 2008; Lafont et al. 2005b) (Fig. 2). CCN3 plays a different role in murine costal and articular chondrocytes, undergoing intramembranous ossification, where it increases proteoglycan synthesis (Janune et al. 2011a).
Far less is known about CCN5 and CCN6 during chondrogenesis. CCN5 overexpression in primary chondrocytes reduces cell viability and PPARγ activation as observed in developmental dysplasia of the hips (DDH) (Ji et al. 2020) (Fig. 2). CCN6 expression increases in adult tissues suggesting a role in cartilage maintenance (Sen et al. 2004): it induces type II collagen and Sox-9 expression and weakly binds and antagonizes IGF-1 induced hypertrophy a collagen type X and Runx2 expression and ALP activity (Cui et al. 2007; Repudi et al. 2013; Sen et al. 2004) (Fig. 2). Loss of CCN6 function, due to at least 70 known mutations, causes progressive pseudo rheumatoid dysplasia (PPD), a rare autosomal disease characterized by symmetrical polyarticular involvement without systemic inflammation, knobbly interphalangeal joints of the hands, and gait abnormalities (for a review see (Torreggiani et al. 2019)). The role of CCN6 in disease pathogenesis is largely unknown, also due to the lack of skeletal phenotype in CCN6 knockout mice (Kutz et al. 2005).
Osteogenesis and fracture healing
All CCN family members were detected in murine bone tissues, with CCN6 being the least expressed protein: CCN1, CCN2, CCN3 and CCN6 were observed only on bone surfaces, whereas CCN4 and CCN5 were detected in bone matrix as well (Kawaki et al. 2011). CCN1, CCN2, CCN4 expression peaks at days 14–28 during experimental osteogenesis in vitro, whereas CCN3 expression steadily decreases. CCN5 expression slightly increases during osteogenesis in vitro; exogenous CCN5 has no effect on osteoblast proliferation, but induces osteoblast maturation by upregulating Osterix, ALP and Bsp via Smad 1/5/8, β-catenin and p38 mediated pathways (Kawaki et al. 2011) (Fig. 3). The expression of CCN6 is very low during osteogenesis and its exogenous administration or silencing has no effect on osteogenesis in vitro (Kawaki et al. 2011).

Schematic representation of the role of different CCN proteins in osteogenesis
CCN1 and CCN2 promote proliferation and differentiation inducing the expression of type I collagen, Runx2 and Bsp via Smad 1/5/8, β-catenin and p38 mediated pathways (Kawaki et al. 2011) (Fig. 3). Osteoblast-specific CCN1 deletion in CCN1OCN mice leads to decreased mineral density, bone volume-to-total volume ratios, trabecular bone surface and area, and decreased peak and fail force (Zhao et al. 2018). As observed for chondrogenesis, CCN2 also plays a more profound role in osteogenesis, with more potent effects on osteoblast proliferation, adhesion, and differentiation and enhancement of ALP expression and activity, osteocalcin, osterix and osteopontin resulting in enhanced formation of mineralized nodules in vitro (Kawaki et al. 2011; Nishida et al. 2000; Safadi et al. 2003; Smerdel-Ramoya et al. 2008; Wang et al. 2009). CCN2 is again regulated in concert with osteogenic factors: CCN2 mediates the osteogenic potential of Notch, preptin and Wnt signalling and its expression is induced by TGFβ, BMP2, IGF-1, cortisol, and taurine in osteoblasts (Maeda et al. 2009; Nakanishi et al. 2000; Pereira et al. 2000; Yuan et al. 2007), antagonizes BMP4 inhibitory action on osteoblasts (Smerdel-Ramoya et al. 2008) and is in turn antagonized by PPARγ and TNFα (Ni et al. 2020; Yu et al. 2014). Upregulation of CCN2 is observed in skeletal pathologies such as ossification of the posterior longitudinal ligament (OPLL) (Yamamoto et al. 2002) and spondylocarpotarsal syntosis (SCT) (Zieba et al. 2016). Consistently, both CCN1 and CCN2 are overexpressed in the first phases of bone healing and in osteoblasts under mechanical stress (Hadjiargyrou et al. 2000; Hu et al. 2012; Kadota et al. 2004; Kanyama et al. 2013; Kulkarni et al. 2012; Lienau et al. 2006). CCN4 plays a more limited role in osteogenesis; it has no osteogenic potential per se, but directly binds BMP2 and promotes its binding to integrin receptors in MSCs with a synergistic effect on BMP-promoted osteogenesis (Inkson et al. 2009; Kawaki et al. 2011) (Fig. 3). CCN4 expression is induced in temporary calluses in structures linking new to old bone upon injury (French et al. 2004) and in human preosteoblastic CIMC-4 cell line upon uniform biaxal 2% strain (Case et al. 2008). Consistent with a pro-osteogenic role, CCN4 knock in mice display denser bone in females, and CCN4-/- mice fail to repair fractures (French et al. 2004).
As observed for chondrogenesis, CCN3 has opposite effects to CCN2 and inhibits osteogenesis by directly binding to BMP2 and inhibiting its downstream signalling (Kawaki et al. 2011; Minamizato et al. 2007; Rydziel et al. 2007) (Fig. 3). Consistently, mice overexpressing CCN3 in osteoblasts, under the osteocalcin promoter, show decreased mineral density and bone formation rates (Rydziel et al. 2007). CCN3 expression increases upon drilled hole femoral injury in osteogenic cells (Matsushita et al. 2013).
Osteoclastogenesis
Knowledge concerning CCN family members in osteoclastogenesis is limited to CCN1 and CCN2, that play contrasting roles (Fig. 4).

Schematic representation of the role of different CCN proteins in osteoclastogenesis
CCN1 inhibits osteoclastogenesis in murine, rabbit and human macrophages stimulated with M-CSF and RANKL; it decreases TRAP expression and activity as well as the expression of integrins, calcitonin receptors, cathepsin K and MMP-9. The effects of CCN1 are not mediated by induction of apoptosis, inhibited cell fusion, or alterations in the p38, ERK and NF-kB pathways. Moreover, the number and activity of mature osteoclasts are not repressed by CCN1 (Crockett et al. 2007).
CCN2 has no osteoclastogenic potential per se, but it favours RANKL-induced osteoclastogenesis in RAW 264.7 macrophages with pleiotropic activities. It synergizes with RANKL in inducing NF-kB translocation into nuclei, activating p38 and inducing NFATC1 and c-Fos; it inhibits Bcl6, an osteoclastogenesis repressor, and favours cell fusion via DC-STAMP (Aoyama et al. 2015; Nishida et al. 2011, 2019) and it counteracts anti-osteoclastogenic effect of osteoprotegerin (Aoyama et al. 2015). The pivotal role of CCN2 in osteoclastogenesis is further corroborated by downregulation of miR-26a, which would inhibit CCN2 itself (Kim et al. 2015).
Consistently, CCN1OCN and CCN2 overexpressing mice show an increased number and activity of osteoclasts, together with upregulation of RANKL and inhibition of osteoprotegerin (Zhao et al. 2018). In contrast, fewer osteoclasts are identifiable in the bones of CCN2-/- mice and stimulation with RANKL and M-CSF in foetal liver cells fails to induce osteoclastogenesis (Nishida et al. 2011).
Myogenesis
All CCN family members, except CCN5 and CCN6, were found to be expressed during myogenesis; for a detailed description of the role of CCN proteins in the physiology of the skeletal muscle the reader is also referred to (Rebolledo et al. 2021).
CCN1 is expressed at all stages of myogenesis in vitro and in vivo under the control of NFATC5, which in turn fosters myoblast migration and differentiation (O’Connor et al. 2007) (Fig. 5). Similarly, upon induction by TGFβ1 and LPA, CCN2 stimulates myoblasts proliferation, upregulates fibronectin, integrins α4, α5, α6 and β1 and MyoD but represses markers of terminal differentiation of myocytes (Nishida et al. 2015) (Fig. 5). CCN1 and CCN2 expression is also up regulated in human skeletal muscles of individuals following a jumping exercise (Kivelä et al. 2007) and downregulated following muscle denervation (Magnusson et al. 2005), thus indicating a possible role in physiologically working muscles.

Schematic representation of the role of different CCN proteins in myogenesis
CCN4 also displays myogenic activity; its expression increases as myoblasts differentiate and its inhibition decreases the expression of myosin heavy chain, myogenin, MRF and MEF-2 (Wang et al. 2008) (Fig. 5). Consistently, CCN4 knockout mice display lower expression of myogenic markers and reduced myofiber repair; moreover, aged osteoblasts expressing less CCN4 fail to sustain muscle stem cell proliferation and differentiation (van den Bosch et al. 2017).
CCN3 is reported to mediate myoblast adhesion in a calcium and integrin-dependent manner and to augment myoblast sensitivity to IGF-1, FGF2 and PDGF (Lafont et al. 2005a) but its overexpression in C2/4 cells hinders myotube formation (Sakamoto et al. 2002) (Fig. 5).
CCN proteins in degenerative and inflammatory diseases of the musculoskeletal system
Degenerative and inflammatory diseases of the musculoskeletal system severely limit patients' quality of life and have high medical costs. CCN family member deregulation has been identified in both osteoarthritis and rheumatoid arthritis and offers therapeutic insights (for a review see (MacDonald et al. 2021)). Osteoarthritis (OA) is an age-related degenerative disease of the articular cartilage, that progressively becomes eroded and substituted by fibrotic tissue (for a review see (Martel-Pelletier et al. 2016)). Rheumatoid arthritis (RA) is an inflammatory disease of joints. The synovium is infiltrated by inflammatory cells, which cause oedema, hyperplasia of the synovial lining, and degradation of the ECM (for a review see (Smolen et al. 2018)). CCN2 has also been ascribed a central role in intervertebral disc disease (IVD) which is the main cause of low back pain in adults (Matta et al. 2018; Wang et al. 2018): CCN2 downregulation associated with aging (Hyiama et al. 2018) and CCN2 notochord-specific knockout leads to precocious and more pronounced degeneration (Bedore et al. 2013).
Moreover, CCN2 is found in the cytoplasm of necrotic and regenerating fibres and in active fibroblasts in the endomysial space of dystrophic human muscles and in mdx mice affected by Duchenne muscular dystrophy (DMD) (Morales et al. 2018; Song et al. 2017; Sun et al. 2008). CCN2 overexpression has proven detrimental for muscle fibres as it transiently augments the expression of fibronectin, collagen type III and decorin and diminishes isometric specific and tetanic forces as well as the response to BaCl2 damage (Morales et al. 2011). Consistently, genetic CCN2 inactivation in mdx mice increases the running ability of mice, reducing necrosis, myogenin and myosin heavy chain (MHC) expression, and collagen stiffness via downregulation of Lox (Petrosino et al. 2019); also, the effects of ACE inhibitors on dystrophy are mediated by a decrease in CCN2 expression (Morales et al. 2013). For further insights into the role of CCN proteins in pathology and regeneration of the skeletal muscle, the reader is also referred to (Rebolledo et al. 2021).
Osteoarthritis
CCN family members have been found to be differentially expressed in the superficial cartilage layer and matrix of OA patients, with maximal expression levels for CCN2, moderate expression levels for CCN1 and CCN4, low expression level for CCN3 and CCN5 and no expression for CCN6 (Komatsu et al. 2015).
CCN1 and CCN2 concentrations in sera and/or synovial fluid are positively correlated with disease severity (Chijiiwa et al. 2015; Honsawek et al. 2012; Komatsu et al. 2015). Monoiodoacetate intra-articular (MIA)- injected mice, an experimental model of OA, also show upregulation of CCN2 expression (Nishida et al. 2004). The role of CCN1 in OA pathogenesis is mainly related to the promotion of inflammatory response as it is induced upon TGFβ, TNFα and IL1β stimulation in osteoarthritic chondrocytes and HCS-2/8 cells (Chijiiwa et al. 2015; Moritani et al. 2005). CCN2 participates in OA pathogenesis upregulating IL1β and VEGF expression through inhibition of miR-210 (Liu et al. 2015b) and the establishment an autocrine loop with M-CSF fostering osteoclastogenesis (Nakao et al. 2005). The role of CCN2 in OA is corroborated by elevated expression of lncRNA PVTI targeting miR-26b, which would in turn inhibit CCN2 (Ding et al. 2020). OA is ameliorated by CCN1 inhibition through siRNA in a model of gouty arthritis induced by injection of monosodium urate crystals or through berberine in a collagen-induced OA (CIOA) murine model via downregulation of IL6, TNFα and IL1β expression via NF-kB pathway (Zhou et al. 2020). On the contrary, OA is ameliorated by additional CCN2 expression, which promotes cartilage repair; local administration via a CCN2-loaded hydrogel fosters reparation of articular cartilage through upregulation of tenascin-C, aggrecan and type X collagen expression (Nishida et al. 2004), and the beneficial effects of hyaluronan are mediated by induction of CCN2 expression in human synovial cells (Lee et al. 2010b). Consistently, the appearance of osteoarthritic-like degeneration is delayed up to 21 weeks in CCN2 knock-in mice: increased cellularity and decreased proteoglycan loss are derived from reduced collagen type X and MMP13 and collagen type I expression in articular chondrocytes (Itoh et al. 2013). Similarly, CCN4 is up regulated in synovial tissues fluid of OA patients and the damaged cartilage of OA experimental models (van den Bosch et al. 2019). CCN4 polymorphisms are associated with spinal OA in Japanese women (Urano et al. 2007). Detrimental effects of CCN4 overexpression include both direct effects on cartilage such as decreased matrix component synthesis and deposition, increased matrix degradation, due to augmented expression and activity of aggrecanase and metalloproteinase, and proinflammatory effects such as upregulation of IL6 and monocyte adhesion through upregulation of VCAM-1 expression (Hou et al. 2013; van den Bosch et al. 2015, 2019).
CCN3 expression shows a positive correlation with age in both mice and humans (Kuwahara et al. 2020). The role of CCN3 in cartilage degeneration is cumbersome; both mice overexpressing CCN3 and mice with targeted disruption of CCN3 show precocious degenerative changes (Kuwahara et al. 2020; Roddy and Boulter 2015). Findings in osteoarthritis confirm this dualism. Indeed, CCN3 expression leads to p53 accumulation in nuclei and p21 promoter activation and hence correlates with the expression of senescence markers, together with the expression of proinflammatory cytokines IL-6 and IL-8 and decreased expression of aggrecan (Kuwahara et al. 2020). On the other hand, CCN3 protects articular cartilage from damage as it antagonizes IL1β-induced upregulation of aggrecanase, MMPs, and iNOS expression and induction of autophagy in chondrocytes by blocking HMGB1 and TLR4. Nevertheless, IL1β has itself been shown to be capable of diminishing CCN3 expression in both SW1353 cells and rat primary chondrocytes (Huang et al. 2019).
Rheumatoid arthritis
All CCN family members were found to be up regulated in RA sera and tissues (Komatsu et al. 2015), but only the role of CCN1 and CCN2 in its pathogenesis has been fully explored. CCN3 expression correlates with disease activity and IL6 production (Wei et al. 2020) and CCN4 single nucleotide polymorphisms have been associated with the risk of developing RA and response to therapy (Kuo et al. 2019).
Both CCN1 and CCN2 are up regulated in synovial tissues, fibroblast-like synoviocytes and sera of RA patients (Chen et al. 2017a, b; Ding et al. 2016; Jie et al. 2015; Nozawa et al. 2009; Zhai et al. 2017; Sun et al. 2020; Zhu et al. 2015). A CCN1 serum concentration higher of 99.66 pg/ml (Fan et al. 2019) and a CCN2 higher than 73.35 pg/ml (Yang et al. 2017) correlate with disease. CCN1 and CCN2 have similar roles in RA pathogenesis, as both favour fibroblast-like synoviocite proliferation and induce osteoclastogenesis as well as angiogenesis (Chen et al. 2017a, 2017b; Ding et al. 2016; Ganesan and Rasool 2017; Nozawa et al. 2009; Zhang et al. 2009). Moreover, CCN1 induces the expression of the proinflammatory cytokine IL-6 (Choi et al. 2020; Lin et al. 2012), IL8 and CCL20 which promote neutrophil and monocyte migration to the synovium (Chen et al. 2017a; Zhu et al. 2015), and MMPs, which foster cell migration (Kwon et al. 2017).
Given their pleiotropic role in RA pathogenesis, a multitude of efforts have been made to inhibit CCN1 and CCN2 with a great diversity of approaches. Targeted gene editing (lentishCCN1, AdshCCN1) and administration of anti-CCN1 monoclonal antibodies, or drugs (rosiglitazone) in collagen-induced (CIA) murine models of RA invariably led to an amelioration of RA symptoms with decreased inflammatory scores and monocyte infiltration, decreased production of proinflammatory cytokines such as IL-6 and IL1β and increased production of antinflammatory IL10, decreased MMP activity and osteoclastogenesis with reduced RORc, TNFα, IL17 and sRANKL levels, and ultimately reduced osteoclast populations (Chen et al. 2017a, b; Kwon et al. 2017; Lin et al. 2012; Zhai et al. 2017; Zhu et al. 2015). Blocking pathways upstream of CCN1 induction, such as IL17 via cyanidin or ferulic acid, or TNFα via SIRT1 or simvastatin, also led to amelioration of RA symptoms (Ganesan and Rasool 2017; Kok et al. 2013; Samarpita et al. 2020). In addition to CCN1, CCN2 targeting by monoclonal antibodies in CIA mice reduced the incidence and symptoms of RA, Th17 population and osteoclastogenesis, T cell proliferation and MMP3 activity (Nozawa et al. 2013; Nozawa 2014).
CCN proteins in sarcomas of the musculoskeletal system
CCN proteins actively participate in carcinogenesis, as their role has been clarified in every hallmark of cancer except inflammation (for a review see (Yeger and Perbal 2021)). Nevertheless, the role of at least CCN1 and CCN2 in rheumatoid arthritis underscores their association with the regulation of proinflammatory cytokines in the microenvironment. Thus, CCN proteins are involved in cancer in a pleiotropic manner and particularly serve sarcomas of the musculoskeletal system.
Osteosarcoma
Osteosarcoma is the most frequent malignancy of bone and commonly affects children and young adults. CCN1, CCN2, CCN3 and CCN4 are all poorly expressed in healthy adult bony tissues, but they are up regulated in primary samples as well as osteosarcoma cell lines (Chen et al. 2013a; Fromigue et al. 2011; Habel et al. 2019; Huang et al. 2016; Liu et al. 2017b; Perbal et al. 2008; Tsai et al. 2014a, 2014b, 2014c; Wu et al. 2013; Zhang et al. 2013). The pivotal roles of CCN1 and CCN2 in osteosarcoma are corroborated by the decreased expression of miRNAs inhibiting their expression, such as miR-33a, miR-100 and miR-365, and miR-584, respectively (Huang et al. 2014, 2018; Li et al. 2020; Xu et al. 2018); on the other hand, miR-25-3p which indirectly fosters CCN1 and CCN2 expression is up regulated in osteosarcoma (Rao et al. 2020). CCN1 and CCN2 do not promote osteosarcoma cell proliferation per se but protect cells from apoptosis and decrease chemotherapeutic efficacy (Chen et al. 2013a; Fromigue et al. 2011; Habel et al. 2015, 2019; Huang et al. 2016; Tsai et al. 2014a, 2014b, 2014c; Zhang et al. 2013). A plethora of in vitro and preclinal models have ascertained the roles of CCN1 and CCN2 in promoting migration and invasiveness. CCN1 induces MMP-2, MMP-9, MMP-14 and TIMP-3, mediates prometastatic effects via the IGF-1/IGF1Rβ pathway, and is elevated in MG63 M7 and M10 sublines selected in vivo for a migratory phenotype; in addition, its inhibition decreases lung-metastatic potential of osteosarcoma cell lines in vivo (Chen et al. 2013a; Habel et al. 2015, 2019; Hou et al. 2014). CCN2 promotes osteosarcoma cell migration by downregulating miR-519d and upregulating MMP-2 and MMP-3 (Hou et al. 2018; Tsai et al. 2014c). CCN4 also promotes migration, invasiveness and metastasis of osteosarcoma cells upregulating MMP-2 and MMP-9 expression (Tsai et al. 2017; Wu et al. 2013). Moreover, CCN1 and CCN4 have proangiogenic activity promoting expression of VEGFA (Habel et al. 2015; Kim et al. 2018; Liu et al. 2017b). Last, CCN1 promotes epithelial-to-mesenchymal transition (EMT) skewing cells towards a spindle-like morphology and a more staminal and migratory phenotype (Habel et al. 2019; Hou et al. 2014). CCN3 deserves separate mention as its role in osteosarcoma is controversial. On the one hand, consistent with the antiproliferative role in osteocytes, its upregulation in 143B cells with poor CCN3 expression decreases cell vitality, proliferation and clonogenicity, and its silencing in MG63 cells yields the opposite effect (Huang et al. 2011). On the other hand, CCN3 has been associated with increased migration and upregulation of COX-2 expression (Huang et al. 2011; Yao et al. 2015) and CCN3 expression correlates with poor prognosis, high expression of MRP1-4 and resistance to cisplatin, doxorubicin, and methotrexate (Perbal et al. 2008).
Chondrosarcoma
Chondrosarcoma is the second most common malignancy of bone and is characterized by poor response to therapy and metastatic phenotype. CCN1, CCN2, CCN3 and CCN4 are expressed in enchondromas and low-grade chondrosarcomas, whereas their expression diminishes as disease progresses to higher grades and less differentiated lesions (Hou et al. 2011; Shakunaga et al. 2000; Tan et al. 2009a, 2009b; Yu et al. 2003). CCN6 expression is also up regulated in chondrosarcoma with respect to healthy tissues and its expression correlates with a metastatic phenotype (Fong et al. 2012). Consistent with their role in osteosarcoma, CCN proteins expression foster migration in chondrosarcoma by augmenting expression and enzymatic activity of MMP-13 (Tan et al. 2009a, 2009b; Tzeng et al. 2011), MMP-2 (Hou et al. 2011) or ICAM-1 (Fong et al. 2012).
Ewing sarcoma
Ewing sarcoma is an aggressive malignancy of bone that often develops in children, molecularly characterized by translocation most commonly involving the EWS and FLI1 genes. CCN1, CCN2 and CCN3 are expressed in all Ewing cell lines, though heterogeneously (Perbal et al. 2009; Strammiello et al. 2003). As observed in osteosarcoma, the role of CCN3 in Ewing sarcoma is controversial. Its exogenous expression in TC-71 cells blocks proliferation, clonogenicity and the cell cycle, increases apoptosis and decreases tumorigenic potential in vivo (Benini et al. 2005). Nevertheless, as observed in osteo- and chondrosarcoma, CCN3 promotes TC-71 cell migration and invasion, decreasing adhesion on collagen type I and IV via integrin α2β1 (Benini et al. 2005). Consistently, CCN3 expression correlates with higher metastatic potential in clinical samples (Perbal et al. 2009).
Rhabdomyosarcoma
Rhabdomyosarcoma is a malignancy of skeletal muscles and represents the most common soft tissue cancer in infancy. CCN2 is expressed in spontaneous rhabdomyosarcomas of the genitourinary tract of Balbp53neu transgenic mice (Croci et al. 2007) and human rhabdomyosarcoma cell lines of both alveolar and embryonal subtypes (Croci et al. 2004). CCN2 inhibition hinders growth and induces apoptosis, though diminishing myogenic differentiation, in accordance with the role of CCN2 in myogenesis (Croci et al. 2004).
Bone metastasis of other cancers
Bone is a preferential site of metastasis for breast cancer (70%) and prostate cancer (85%) and myeloma (95%) (Coleman et al. 2020). Metastatic dissemination of tumour cells requires adaptation to the unique milieu of specialized bone cells, mineralized bone matrix: CCN proteins, as cellular communication networks, mediate this heterologous interaction, thus generally favouring bone invasion and metastatic cells proliferation with the exceptions CCN6 in breast cancer and CCN1 in myeloma (Fig. 6). In this field, the most studied family member is CCN2 which also figures in Kang’s bone metastasis signature for genes cooperating in bone metastasis development irrespective of the primary tumour site (Casimiro et al. 2012). In addition to frequent bone-metastatic cancer, discussed below, CCN2 is highly expressed in bone invasive oral squamous carcinoma (Shimo et al. 2008), melanoma (Braig et al. 2011) and bone metastasis of hepatocellular carcinoma in both murine models and clinical samples (Hou et al. 2015; Xiang et al. 2011, 2012), where high CCN2 expression positively correlates with rate of bone metastasis (Xiang et al. 2011; Zhang et al. 2019).

Schematic representation of the role of different CCN proteins in bone metastasis of other cancers
Breast cancer
CCN1 and CCN2 are up regulated in breast cancer and their expression correlates with staging and local (CCN1) or bone (CCN2) invasiveness (Kim et al. 2020; O’Kelly et al. 2008). Forced CCN1 expression in MCF7 low-invasive breast cancer cell line increases its ability to adhere to collagen and fibronectin, invasiveness on fibronectin substrate and migratory ability (O’Kelly et al. 2008). Comparison of different breast cancer cell lines or parental cells with those experimentally enriched for metastatic potential, invariably highlighted overexpression of CCN2 (Kim et al. 2020; Ohgawara et al. 2011). CCN2 cooperates in angiogenesis and osteoclastogenesis induced by metastatic breast cancer cells, inducing angiopoietin 2 and RANKL (Kim et al. 2020). Consistently, its silencing diminishes invasive potential, MMP-9 and MMP-13 expression and activity, and Runx2 expression in vitro (Ren et al. 2014) and an anti-CCN2 monoclonal antibody reduces metastatic potential of MDA-MB-231 upon intracardiac injection with remaining metastases displaying a less angiogenetic and osteoclastic phenotype (Shimo et al. 2006). In contrast with the prometastatic role of CCN1 and CCN2, CCN6 acts as an oncosuppressor in breast cancer, preserving epithelial differentiation. CCN6 silencing in human mammary epithelial cells (HME) leads to loss of acinar polarity, clear cell–cell borders, decreased E-cadherin and integrin α6 expression and failure to form a lumen, thus favouring a less epithelial and more invasive phenotype. Conversely, forced CCN6 expression in MDA-MD-231 and SUM149 cells reverts the migratory phenotype and restores the ability to form an acinar lumen. Consistently, CCN6 expression is downregulated in human breast cancer (Pal et al. 2012); for a review of CCN6 role in breast cancer see (Tran and Kleer 2018).
Prostate cancer
CCN2, as observed in breast cancer, CCN3 and CCN4 levels are up regulated in prostate cancer and bone-metastatic cells or corresponding bone metastasis (Dankner et al. 2019; Kim et al. 2020; Ono et al. 2013). Consistently, exogenous administration of CCN2 prior to in vivo injection increases number and size of bone metastases, whereas its inhibition reduces bone metastasis and related osteosclerosis (Kim et al. 2020; Zhang et al. 2018). CCN3 in conditioned medium from prostate cancer cells favours osteoclastogenesis via both RANKL-dependent and RANKL-independent pathways in vitro and forced overexpression of CCN3 in PCa and LnCAP C4-2 cells enhances osteolytic potential and TRAP-staining in metastatic lesions (Chen et al. 2013b). CCN4 promotes adhesion of prostate cancer cells to bone via upregulation of VCAM1 and integrin α4β1 expression on osteoblasts and migration of prostate cancer cells themselves (Chang et al. 2018; Tai et al. 2014). Inhibition of CCN4 by monoclonal antibodies reduces metastatic burden and in particular metastases to bone versus other localizations reducing migration and invasiveness (Ono et al. 2013).
Myeloma
CCN1 is up regulated in multiple myeloma tissues as well as in patients’ sera (Johnson et al. 2014; Liu et al. 2017a). Contrary to what observed for other CCN proteins and cancers, its expression positively correlates with better prognosis; consistent with its inhibitory role in osteoclastogenesis, exogenous administration of CCN1 to multiple myeloma cells in coculture with MSCs slows proliferation and bone resorption, favouring osteoblasts to the detriment of osteoclasts (Johnson et al. 2014; Liu et al. 2017a).
Concluding remarks
CCN1 and CCN2 play pivotal stimulating roles in development, physiology, pathology, and malignancy of the musculoskeletal system. CCN4 has a minor role in all aspects whereas CCN3 counteracts the actions of CCN2. The roles of CCN5 and CCN6 in the musculoskeletal system remain poorly studied and unclear. Efforts should be made to perform a more coordinated study of the effects of these family members as a team rather than single entities in the physiology of both osteoclasts and skeletal muscle cells as well as degenerative diseases and cancer. Regenerative approaches could also benefit from considering the integrated role of CCN family members in fine-tuning the function of diverse tissues and cell types in the musculoskeletal system. Lessons learned from the proinflammatory role of CCN1 and CCN2 in rheumatoid arthritis should prompt studies on the ability of CCN family members to affect the tumour microenvironment and immune evasion. Differential expression between osteosarcomas/bone metastasis and surrounding normal bone or between low- and high-grade chondrosarcomas offers a rationale for developing targeted therapeutics, at least for young adult patients. Last, given the involvement of CCN2 in bone metastasis regardless of the of primary tumour site, its level in biological fluid could provide a precocious marker of skeleton related events.
Abbreviations
- CCN1-6:
-
Cellular communication network 1–6
- ECM:
-
Extra-cellular matrix
- mRNA:
-
Messenger RiboNucleic Acid
- BMP-2, 3, 4, 12:
-
Bone morphogenetic protein 2, 3, 4, 12
- TGFß1-3:
-
Transforming growth factor ß 1–3
- IL1β, 6, 8, 10, 17:
-
Interleukin 1β, 6, 8, 10, 17
- Runx 2:
-
Runt-related transcription factor 2
- α-SMA:
-
Smooth muscle α -actin
- MyoD:
-
Myogenic differentiation antigen
- Myf5:
-
Myogenic Factor 5
- MRF:
-
Myogenic regulatory factors
- MHC:
-
Myosin heavy chain
- DNA:
-
DesossicriboNucleic Acid
- Sox-9:
-
SRY-Box Transcription Factor 9
- ALP:
-
Alkaline phosphatase
- M-CSF:
-
Macrophage colony-stimulating factor
- FGF 1, 2:
-
Fibroblast Growth Factor 1‚ 2
- Smad 1/2/3/5/8:
-
Small mothers against decapentaplegic 1/2/3/5/8
- Wnt3a:
-
Wingless/Integrated 3a
- PPARγ:
-
Peroxisome Proliferator Activated Receptor γ
- IGF:
-
Insulin-Like Growth Factor 1
- Bsp:
-
Bone Sialoprotein
- p38:
-
Mitogen-activated protein kinases
- TNF α:
-
Transforming growth factor α
- (s)RANKL (soluble):
-
Receptor Activator of Nuclear Factor Kappa B Ligand
- NF-kB:
-
Nuclear Factor Kappa Beta
- NFATC1, 5:
-
Nuclear factor of activated T cells 1, 5
- Bcl6:
-
B-cell lymphoma 6 protein
- TRAP:
-
Tartrate-Resistant Acid Phosphatase
- MMP 1, 2, 3, 9, 3, 14:
-
Matrix metalloproteinase 1, 2, 3, 9, 13, 14
- ERK:
-
Extracellular Signal-Regulated Kinase
- DC-STAMP:
-
Dendritic Cell-specific Transmembrane Protein Allergy
- MEF-2:
-
Myocyte enhancer factor 2
- PDGF:
-
Platelet-derived growth factor
- Lox:
-
Lectin-like oxidized low-density lipoprotein (LDL) receptor
- ACE:
-
Angiotensin Converting Enzyme
- VEGF:
-
Vascular endothelial growth factor
- lncRNA:
-
Long non coding RiboNucleic Acid
- PVT1:
-
Human Plasmacytoma Variant Translocation 1
- siRNA:
-
Silencing RiboNucleic Acid
- VCAM-1:
-
Vascular cell adhesion molecule 1
- P21:
-
Cyclin-dependent kinase inhibitor 21
- iNOS:
-
Inducible Nitric Oxide Synthase
- HMGB1:
-
High mobility group box 1
- TLR4:
-
Toll-like receptor 4
- CCL20:
-
Chemokine (C–C motif) ligand 20
- RORc:
-
RAR related orphan receptor C
- SIRT1:
-
Sirtuin (silent mating type information regulation 2 homolog) 1
- TIMP-3:
-
Metalloproteinase inhibitor 3
- IGF1Rβ:
-
Insulin like growth factor 1 receptor β
- MRP1-4:
-
Multidrug resistance-associated protein 1–4
- ICAM-1:
-
Intercellular Adhesion Molecule 1
References
Aoyama E, Kubota S, Khattab HM, Nishida T, Takigawa M (2015) CCN2 enhances RANKL-induced osteoclast differentiation via direct binding to RANK and OPG. Bone 73:242–248. https://doi.org/10.1016/j.bone.2014.12.058
Bedore J, Sha W, McCann MR, Liu S, Leask A, Séguin CA (2013) Impaired intervertebral disc development and premature disc degeneration in mice with notochord-specific deletion of CCN2. Arthritis Rheum 65:2634–2644. https://doi.org/10.1002/art.38075
Benini S, Perbal B, Zambelli D, Colombo MP, Manara MC, Serra M, Parenza M, Martinez V, Picci P, Scotlandi K (2005) In Ewing’s sarcoma CCN3(NOV) inhibits proliferation while promoting migration and invasion of the same cell type. Oncogene 24:4349–4361. https://doi.org/10.1038/sj.onc.1208620
Braig S, Wallner S, Junglas B, Fuchshofer R, Bosserhoff A-K (2011) CTGF is overexpressed in malignant melanoma and promotes cell invasion and migration. Br J Cancer 105:231–238. https://doi.org/10.1038/bjc.2011.226
Case N, Ma M, Sen B, Xie Z, Gross TS, Rubin J (2008) Beta-catenin levels influence rapid mechanical responses in osteoblasts. J Biol Chem 283:29196–29205. https://doi.org/10.1074/jbc.M801907200
Casimiro S, Luis I, Fernandes A, Pires R, Pinto A, Gouveia AG, Francisco AF, Portela J, Correia L, Costa L (2012) Analysis of a bone metastasis gene expression signature in patients with bone metastasis from solid tumors. Clin Exp Metas 29:155–164. https://doi.org/10.1007/s10585-011-9438-0
Cernea M, Tang W, Guan H, Yang K (2016) Wisp1 mediates Bmp3-stimulated mesenchymal stem cell proliferation. J Mol Endocrinol 56:39–46. https://doi.org/10.1530/JME-15-0217
Chal J, Pourquié O (2017) Making muscle: skeletal myogenesis. Development 144:2104–2122. https://doi.org/10.1242/dev.151035
Chang A-C, Chen P-C, Lin Y-F, Su C-M, Liu J-F, Lin T-H, Chuang S-M, Tang C-H (2018) Osteoblast-secreted WISP-1 promotes adherence of prostate cancer cells to bone via the VCAM-1/integrin α4β1 system. Cancer Lett 426:47–56. https://doi.org/10.1016/j.canlet.2018.03.050
Chen C-Y, Fuh L-J, Huang C-C, Hsu C-J, Su C-M, Liu S-C, Lin Y-M, Tang C-H (2017a) Enhancement of CCL2 expression and monocyte migration by CCN1 in osteoblasts through inhibiting miR-518a-5p: implication of rheumatoid arthritis therapy. Sci Rep 7:421. https://doi.org/10.1038/s41598-017-00513-0
Chen C-Y, Su C-M, Hsu C-J, Huang C-C, Wang S-W, Liu S-C, Chen W-C, Fuh L-J, Tang C-H (2017b) CCN1 promotes VEGF production in osteoblasts and induces endothelial progenitor cell angiogenesis by inhibiting miR-126 expression in rheumatoid arthritis: CCN1 INCREASES VEGF EXPRESSION IN OSTEOBLASTS. J Bone Miner Res 32:34–45. https://doi.org/10.1002/jbmr.2926
Chen J, Song Y, Yang J, Gong L, Zhao P, Zhang Y, Su H (2013a) The up-regulation of cysteine-rich protein 61 induced by transforming growth factor beta enhances osteosarcoma cell migration. Mol Cell Biochem 384:269–277. https://doi.org/10.1007/s11010-013-1807-3
Chen P-C, Cheng H-C, Tang C-H (2013b) CCN3 promotes prostate cancer bone metastasis by modulating the tumor–bone microenvironment through RANKL-dependent pathway. Carcinogenesis 34:1669–1679. https://doi.org/10.1093/carcin/bgt103
Chijiiwa M, Mochizuki S, Kimura T, Abe H, Tanaka Y, Fujii Y, Shimizu H, Enomoto H, Toyama Y, Okada Y (2015) CCN1 (Cyr61) is overexpressed in human osteoarthritic cartilage and inhibits ADAMTS-4 (Aggrecanase 1) activity: CCN1 IN OSTEOARTHRITIC CARTILAGE. Arthr Rheumatol 67:1557–1567. https://doi.org/10.1002/art.39078
Choi C, Jeong W, Ghang B, Park Y, Hyun C, Cho M, Kim J (2020) Cyr61 synthesis is induced by interleukin-6 and promotes migration and invasion of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther 22:275. https://doi.org/10.1186/s13075-020-02369-8
Chong HC, Tan CK, Huang R-L, Tan NS (2012) Matricellular proteins: a sticky affair with cancers. J Oncol 2012:1–17. https://doi.org/10.1155/2012/351089
Coleman RE, Croucher PI, Padhani AR, Clézardin P, Chow E, Fallon M, Guise T, Colangeli S, Capanna R, Costa L (2020) Bone Metastases. Nat Rev Dis Primers 6:83. https://doi.org/10.1038/s41572-020-00216-3
Croci S, Landuzzi L, Astolfi A, Nicoletti G, Rosolen A, Sartori F, Follo MY, Oliver N, De Giovanni C, Nanni P, Lollini P-L (2004) Inhibition of connective tissue growth factor (CTGF/CCN2) expression decreases the survival and myogenic differentiation of human rhabdomyosarcoma cells. Can Res 64:1730–1736. https://doi.org/10.1158/0008-5472.CAN-3502-02
Croci S, Landuzzi L, Nicoletti G, Palladini A, Antognoli A, De Giovanni C, Nanni P, Lollini P-L (2007) Expression of connective tissue growth factor (CTGF/CCN2) in a mouse model of rhabdomyosarcomagenesis. Pathol Oncol Res 13:336–339. https://doi.org/10.1007/BF02940313
Crockett JC, Schütze N, Tosh D, Jatzke S, Duthie A, Jakob F, Rogers MJ (2007) The matricellular protein CYR61 inhibits osteoclastogenesis by a mechanism independent of alphavbeta3 and alphavbeta5. Endocrinology 148:5761–5768. https://doi.org/10.1210/en.2007-0473
Cui RR, Huang J, Yi L, Xie H, Zhou HD, Yuan LQ, Wang M, Peng YQ, Luo XH, Liao EY (2007) WISP3 suppresses insulin-like growth factor signaling in human chondrocytes. Mol Cell Endocrinol 279:1–8. https://doi.org/10.1016/j.mce.2007.08.007
Dankner M, Ouellet V, Communal L, Schmitt E, Perkins D, Annis MG, Barrès V, Caron C, Mes-Masson A-M, Saad F, Siegel PM (2019) CCN3/Nephroblastoma overexpressed is a functional mediator of prostate cancer bone metastasis that is associated with poor patient prognosis. Am J Pathol 189:1451–1461. https://doi.org/10.1016/j.ajpath.2019.04.006
Ding L-B, Li Y, Liu G-Y, Li T-H, Li F, Guan J, Wang H-J (2020) Long non-coding RNA PVT1, a molecular sponge of miR-26b, is involved in the progression of hyperglycemia-induced collagen degradation in human chondrocytes by targeting CTGF/TGF-β signal ways. Innate Immun 26:204–214. https://doi.org/10.1177/1753425919881778
Ding S, Duan H, Fang F, Shen H, Xiao W (2016) CTGF promotes articular damage by increased proliferation of fibroblast-like synoviocytes in rheumatoid arthritis. Scand J Rheumatol 45:282–287. https://doi.org/10.3109/03009742.2015.1092581
Djouad F, Delorme B, Maurice M, Bony C, Apparailly F, Louis-Plence P, Canovas F, Charbord P, Noël D, Jorgensen C (2007) Microenvironmental changes during differentiation of mesenchymal stem cells towards chondrocytes. Arthritis Res Ther 9:R33. https://doi.org/10.1186/ar2153
El-Seoudi A, Abd El Kader T, Nishida T, Eguchi T, Aoyama E, Takigawa M, Kubota S (2017) Catabolic effects of FGF-1 on chondrocytes and its possible role in osteoarthritis. J Cell Commun Signal 11:255–263. https://doi.org/10.1007/s12079-017-0384-8
Etich J, Holzer T, Pitzler L, Bluhm B, Brachvogel B (2015) MiR-26a modulates extracellular matrix homeostasis in cartilage. Matrix Biol 43:27–34. https://doi.org/10.1016/j.matbio.2015.02.014
Fan Y, Yang X, Zhao J, Sun X, Xie W, Huang Y, Li G, Hao Y, Zhang Z (2019) Cysteine-rich 61 (Cyr61): a biomarker reflecting disease activity in rheumatoid arthritis. Arthritis Res Ther 21:123. https://doi.org/10.1186/s13075-019-1906-y
Fong Y-C, Lin C-Y, Su Y-C, Chen W-C, Tsai F-J, Tsai C-H, Huang C-Y, Tang C-H (2012) CCN6 enhances ICAM-1 expression and cell motility in human chondrosarcoma cells. J Cell Physiol 227:223–232. https://doi.org/10.1002/jcp.22720
French DM, Kaul RJ, D’Souza AL, Crowley CW, Bao M, Frantz GD, Filvaroff EH, Desnoyers L (2004) WISP-1 is an osteoblastic regulator expressed during skeletal development and fracture repair. Am J Pathol 165:855–867. https://doi.org/10.1016/S0002-9440(10)63348-2
Friedrichsen S, Heuer H, Christ S, Winckler M, Brauer D, Bauer K, Raivich G (2003) CTGF expression during mouse embryonic development. Cell Tissue Res 312:175–188. https://doi.org/10.1007/s00441-003-0712-6
Fromigue O, Hamidouche Z, Vaudin P, Lecanda F, Patino A, Barbry P, Mari B, Marie PJ (2011) CYR61 downregulation reduces osteosarcoma cell invasion, migration, and metastasis. J Bone Miner Res 26:1533–1542. https://doi.org/10.1002/jbmr.343
Fujisawa T, Hattori T, Ono M, Uehara J, Kubota S, Kuboki T, Takigawa M (2008) CCN family 2/connective tissue growth factor (CCN2/CTGF) stimulates proliferation and differentiation of auricular chondrocytes. Osteoarthr Cartil 16:787–795. https://doi.org/10.1016/j.joca.2007.11.001
Furumatsu T, Kanazawa T, Miyake Y, Kubota S, Takigawa M, Ozaki T (2012) Mechanical stretch increases Smad3-dependent CCN2 expression in inner meniscus cells: STRETCH-INDUCED CCN2 IN THE MENISCUS. J Orthop Res 30:1738–1745. https://doi.org/10.1002/jor.22142
Furumatsu T, Matsumoto E, Kanazawa T, Fujii M, Lu Z, Kajiki R, Ozaki T (2013) Tensile strain increases expression of CCN2 and COL2A1 by activating TGF-β-Smad2/3 pathway in chondrocytic cells. J Biomech 46:1508–1515. https://doi.org/10.1016/j.jbiomech.2013.03.028
Ganesan R, Rasool M (2017) Interleukin 17 regulates SHP-2 and IL-17RA/STAT-3 dependent Cyr 61, IL-23 and GM-CSF expression and RANKL mediated osteoclastogenesis by fibroblast-like synoviocytes in rheumatoid arthritis. Mol Immunol 91:134–144. https://doi.org/10.1016/j.molimm.2017.09.003
Grünberg JR, Hammarstedt A, Hedjazifar S, Smith U (2014) The novel secreted adipokine WNT1-inducible signaling pathway protein 2 (WISP2) is a mesenchymal cell activator of canonical WNT. J Biol Chem 289:6899–6907. https://doi.org/10.1074/jbc.M113.511964
Habel N, Vilalta M, Bawa O, Opolon P, Blanco J, Fromigué O (2015) Cyr61 silencing reduces vascularization and dissemination of osteosarcoma tumors. Oncogene 34:3207–3213. https://doi.org/10.1038/onc.2014.232
Habel N, Stefanovska B, Carène D, Patiño-Garcia A, Lecanda F, Fromigué O (2019) CYR61 triggers osteosarcoma metastatic spreading via an IGF1Rβ-dependent EMT-like process. BMC Cancer 19:62. https://doi.org/10.1186/s12885-019-5282-4
Hadjiargyrou M, Ahrens W, Rubin CT (2000) Temporal expression of the chondrogenic and angiogenic growth factor CYR61 during fracture repair. J Bone Miner Res 15:1014–1023. https://doi.org/10.1359/jbmr.2000.15.6.1014
Hall-Glenn F, Aivazi A, Akopyan L, Ong JR, Baxter RR, Benya PD, Goldschmeding R, van Nieuwenhoven FA, Hunziker EB, Lyons KM (2013) CCN2/CTGF is required for matrix organization and to protect growth plate chondrocytes from cellular stress. J Cell Commun Signal 7:219–230. https://doi.org/10.1007/s12079-013-0201-y
Heath E, Tahri D, Andermarcher E, Schofield P, Fleming S, Boulter CA (2008) Abnormal skeletal and cardiac development, cardiomyopathy, muscle atrophy and cataracts in mice with a targeted disruption of the Nov (Ccn3) gene. BMC Dev Biol 8:18. https://doi.org/10.1186/1471-213X-8-18
Honsawek S, Yuktanandana P, Tanavalee A, Chirathaworn C, Anomasiri W, Udomsinprasert W, Saetan N, Suantawee T, Tantavisut S (2012) Plasma and synovial fluid connective tissue growth factor levels are correlated with disease severity in patients with knee osteoarthritis. Biomarkers 17:303–308. https://doi.org/10.3109/1354750X.2012.666676
Hoshijima M, Hattori T, Inoue M, Araki D, Hanagata H, Miyauchi A, Takigawa M (2006) CT domain of CCN2/CTGF directly interacts with fibronectin and enhances cell adhesion of chondrocytes through integrin alpha5beta1. FEBS Lett 580:1376–1382. https://doi.org/10.1016/j.febslet.2006.01.061
Hou C-H, Chiang Y-C, Fong Y-C, Tang C-H (2011) WISP-1 increases MMP-2 expression and cell motility in human chondrosarcoma cells. Biochem Pharmacol 81:1286–1295. https://doi.org/10.1016/j.bcp.2011.03.016
Hou C-H, Tang C-H, Hsu C-J, Hou S-M, Liu J-F (2013) CCN4 induces IL-6 production through αvβ5 receptor, PI3K, Akt, and NF-κB singling pathway in human synovial fibroblasts. Arthritis Res Ther 15:R19. https://doi.org/10.1186/ar4151
Hou C-H, Lin F-L, Hou S-M, Liu J-F (2014) Cyr61 promotes epithelial-mesenchymal transition and tumor metastasis of osteosarcoma by Raf-1/MEK/ERK/Elk-1/TWIST-1 signaling pathway. Mol Cancer 13:236. https://doi.org/10.1186/1476-4598-13-236
Hou C-H, Yang R, Tsao Y-T (2018) Connective tissue growth factor stimulates osteosarcoma cell migration and induces osteosarcoma metastasis by upregulating VCAM-1 expression. Biochem Pharmacol 155:71–81. https://doi.org/10.1016/j.bcp.2018.06.015
Hou R, Wang Y, Liang H, Zhang Z, Liu Z, Zhang B, Zhang B, Chen X (2015) Animal and cellular models of hepatocellular carcinoma bone metastasis: establishment and characterisation. J Cancer Res Clin Oncol 141:1931–1943. https://doi.org/10.1007/s00432-015-1958-6
Hu K, Tao Y, Li J, Liu Z, Zhu X, Wang Z (2019) A comparative genomic and phylogenetic analysis of the origin and evolution of the CCN gene family. Biomed Res Int 2019:8620878. https://doi.org/10.1155/2019/8620878
Hu Y, Yang HF, Li S, Chen JZ, Luo YW, Yang C (2012) Condyle and mandibular bone change after unilateral condylar neck fracture in growing rats. Int J Oral Maxillofac Surg 41:912–921. https://doi.org/10.1016/j.ijom.2011.12.036
Huang C-Y, Lee C-Y, Chen M-Y, Tsai H-C, Hsu H-C, Tang C-H (2011) Nephroblastoma overexpressed gene (NOV) enhances cell motility and COX-2 upregulation of human osteosarcoma involves αvβ5 integrin, ILK and AP-1-dependent pathways. Biochem Pharmacol 81:577–585. https://doi.org/10.1016/j.bcp.2010.12.005
Huang J, Gao K, Lin J, Wang Q (2014) MicroRNA-100 inhibits osteosarcoma cell proliferation by targeting Cyr61. Tumor Biology 35:1095–1100. https://doi.org/10.1007/s13277-013-1146-8
Huang X, Ni B, Mao Z, Xi Y, Chu X, Zhang R, Ma X, You H (2019) NOV/CCN3 induces cartilage protection by inhibiting PI3K/AKT/mTOR pathway. J Cell Mol Med 23:7525–7534. https://doi.org/10.1111/jcmm.14621
Huang Y, Zhao S, Zhang C, Li X (2016) Downregulation of connective tissue growth factor reduces migration and invasiveness of osteosarcoma cells. Mol Med Rep 13:1888–1894. https://doi.org/10.3892/mmr.2015.4701
Huang Y, Zhang J, Shao H, Liu J, Jin M, Chen J, Zhao H (2018) miR-33a mediates the anti-tumor effect of lovastatin in osteosarcoma by targeting CYR61. Cell Physiol Biochem 51:938–948. https://doi.org/10.1159/000495396
Inkson CA, Ono M, Bi Y, Kuznetsov SA, Fisher LW, Young MF (2009) The potential functional interaction of biglycan and WISP-1 in controlling differentiation and proliferation of osteogenic cells. Cells Tissues Organs 189:153–157. https://doi.org/10.1159/000151377
Itoh S, Hattori T, Tomita N, Aoyama E, Yutani Y, Yamashiro T, Takigawa M (2013) CCN family member 2/connective tissue growth factor (CCN2/CTGF) has anti-aging effects that protect articular cartilage from age-related degenerative changes. PLoS ONE 8:e71156. https://doi.org/10.1371/journal.pone.0071156
Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM (2003) Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 130:2779–2791. https://doi.org/10.1242/dev.00505
Janune D, Kubota S, Lazar N, Perbal B, Iida S, Takigawa M (2011a) CCN3-mediated promotion of sulfated proteoglycan synthesis in rat chondrocytes from developing joint heads. J Cell Commun Signal 5:167–171. https://doi.org/10.1007/s12079-011-0135-1
Janune D, Kubota S, Nishida T, Kawaki H, Perbal B, Iida S, Takigawa M (2011b) Novel effects of CCN3 that may direct the differentiation of chondrocytes. FEBS Lett 585:3033–3040. https://doi.org/10.1016/j.febslet.2011.08.024
Janune D, Abd El Kader T, Aoyama E, Nishida T, Tabata Y, Kubota S, Takigawa M (2017) Novel role of CCN3 that maintains the differentiated phenotype of articular cartilage. J Bone Miner Metab 35:582–597. https://doi.org/10.1007/s00774-016-0793-4
Ji X, Liu T, Zhao S, Li J, Li L, Wang E (2020) WISP-2, an up regulated gene in hip cartilage from the DDH model rats, induces chondrocyte apoptosis through PPARγ in vitro. FASEB J 34:4904–4917. https://doi.org/10.1096/fj.201901915R
Jiang J, Zhao G, Lyons KM (2018) Characterization of bone morphology in CCN5/WISP5 knockout mice. J Cell Commun Signal 12:265–270. https://doi.org/10.1007/s12079-018-0457-3
Jie L-G, Huang R-Y, Sun W-F, Wei S, Chu Y-L, Huang Q-C, Du H-Y (2015) Role of cysteine-rich angiogenic inducer 61 in fibroblast-like synovial cell proliferation and invasion in rheumatoid arthritis. Mol Med Rep 11:917–923. https://doi.org/10.3892/mmr.2014.2770
Johnson SK, Stewart JP, Bam R, Qu P, Barlogie B, van Rhee F, Shaughnessy JD, Epstein J, Yaccoby S (2014) CYR61/CCN1 overexpression in the myeloma microenvironment is associated with superior survival and reduced bone disease. Blood 124:2051–2060. https://doi.org/10.1182/blood-2014-02-555813
Jones JA, Gray MR, Oliveira BE, Koch M, Castellot JJ (2007) CCN5 expression in mammals: I. Embryonic and fetal tissues of mouse and human. J Cell Commun Signal 1:127–143. https://doi.org/10.1007/s12079-007-0012-0
Kadota H, Nakanishi T, Asaumi K, Yamaai T, Nakata E, Mitani S, Aoki K, Aiga A, Inoue H, Takigawa M (2004) Expression of connective tissue growth factor/hypertrophic chondrocyte-specific gene product 24 (CTGF/Hcs24/CCN2) during distraction osteogenesis. J Bone Miner Metab 22:293–302. https://doi.org/10.1007/s00774-004-0486-2
Kanyama M, Shimo T, Sugito H, Nagayama M, Kuboki T, Pacifici M, Koyama E (2013) Regulation of CCN2 gene expression and possible roles in developing tooth germs. Arch Oral Biol 58:1659–1666. https://doi.org/10.1016/j.archoralbio.2013.08.010
Katsube K, Sakamoto K, Tamamura Y, Yamaguchi A (2009) Role of CCN, a vertebrate specific gene family, in development. Dev Growth Differ 51:55–67. https://doi.org/10.1111/j.1440-169X.2009.01077.x
Kawaki H, Kubota S, Suzuki A, Lazar N, Yamada T, Matsumura T, Ohgawara T, Maeda T, Perbal B, Lyons KM, Takigawa M (2008) Cooperative regulation of chondrocyte differentiation by CCN2 and CCN3 shown by a comprehensive analysis of the CCN family proteins in cartilage. J Bone Miner Res 23:1751–1764. https://doi.org/10.1359/jbmr.080615
Kawaki H, Kubota S, Suzuki A, Suzuki M, Kohsaka K, Hoshi K, Fujii T, Lazar N, Ohgawara T, Maeda T, Perbal B, Takano-Yamamoto T, Takigawa M (2011) Differential roles of CCN family proteins during osteoblast differentiation: involvement of smad and MAPK signaling pathways. Bone 49:975–989. https://doi.org/10.1016/j.bone.2011.06.033
Kim B, Kim H, Jung S, Moon A, Noh D, Lee ZH, Kim HJ, Kim H (2020) A CTGF-RUNX2-RANKL axis in breast and prostate cancer cells promotes tumor progression in bone. J Bone Miner Res 35:155–166. https://doi.org/10.1002/jbmr.3869
Kim H, Yoo S, Zhou R, Xu A, Bernitz JM, Yuan Y, Gomes AM, Daniel MG, Su J, Demicco EG, Zhu J, Moore KA, Lee D-F, Lemischka IR, Schaniel C (2018) Oncogenic role of SFRP2 in p53-mutant osteosarcoma development via autocrine and paracrine mechanism. Proc Natl Acad Sci 115:E11128–E11137. https://doi.org/10.1073/pnas.1814044115
Kim K, Kim JH, Kim I, Lee J, Seong S, Park YW, Kim N (2015) Micr regulates RANKL induced osteoclast formation. Mol Cells 38:75–80. https://doi.org/10.14348/molcells.2015.2241
Kireeva ML, Latinkić BV, Kolesnikova TV, Chen CC, Yang GP, Abler AS, Lau LF (1997) Cyr61 and Fisp12 are both ECM-associated signaling molecules: activities, metabolism, and localization during development. Exp Cell Res 233:63–77. https://doi.org/10.1006/excr.1997.3548
Kivelä R, Kyröläinen H, Selänne H, Komi PV, Kainulainen H, Vihko V (2007) A single bout of exercise with high mechanical loading induces the expression of Cyr61/CCN1 and CTGF/CCN2 in human skeletal muscle. J Appl Physiol 103:1395–1401. https://doi.org/10.1152/japplphysiol.00531.2007
Kocialkowski S, Yeger H, Kingdom J, Perbal B, Schofield PN (2001) Expression of the human NOV gene in first trimester fetal tissues. Anat Embryol (berl) 203:417–427. https://doi.org/10.1007/s004290100177
Kok S-H, Lin L-D, Hou K-L, Hong C-Y, Chang C-C, Hsiao M, Wang J-H, Lai EH-H, Lin S-K (2013) Simvastatin inhibits cysteine-rich protein 61 expression in rheumatoid arthritis synovial fibroblasts through the regulation of sirtuin-1/FoxO3a signaling. Arthritis Rheum 65:639–649. https://doi.org/10.1002/art.37807
Komatsu M, Nakamura Y, Maruyama M, Abe K, Watanapokasin R, Kato H (2015) Expression profles of human CCN genes in patients with osteoarthritis or rheumatoid arthritis. J Orthop Sci 20:708–716. https://doi.org/10.1007/s00776-015-0727-3
Kulkarni RN, Bakker AD, Everts V, Klein-Nulend J (2012) Mechanical loading prevents the stimulating effect of IL-1β on osteocyte-modulated osteoclastogenesis. Biochem Biophys Res Commun 420:11–16. https://doi.org/10.1016/j.bbrc.2012.02.099
Kuo S-J, Hsua P-W, Chien S-Y, Huang C-C, Hu S-L, Tsai C-H, Su C-M, Tang C-H (2019) Associations between WNT1-inducible signaling pathway protein-1 (WISP-1) genetic polymorphisms and clinical aspects of rheumatoid arthritis among Chinese Han subjects. Medicine 98:e17604. https://doi.org/10.1097/MD.0000000000017604
Kutz WE, Gong Y, Warman ML (2005) WISP3, the gene responsible for the human skeletal disease progressive pseudorheumatoid dysplasia, is not essential for skeletal function in mice. Mol Cell Biol 25:414–421. https://doi.org/10.1128/MCB.25.1.414-421.2005
Kuwahara M, Kadoya K, Kondo S, Fu S, Miyake Y, Ogo A, Ono M, Furumatsu T, Nakata E, Sasaki T, Minagi S, Takigawa M, Kubota S, Hattori T (2020) CCN3 (NOV) drives degradative changes in aging articular cartilage. Int J Mol Sci 21:7556. https://doi.org/10.3390/ijms21207556
Kwon E-J, Park E-J, Choi S, Kim S-R, Cho M, Kim J (2017) PPARγ agonist rosiglitazone inhibits migration and invasion by downregulating Cyr61 in rheumatoid arthritis fibroblast-like synoviocytes. Int J Rheum Dis 20:1499–1509. https://doi.org/10.1111/1756-185X.12913
Lafont J, Thibout H, Dubois C, Laurent M, Martinerie C (2005a) NOV/CCN3 induces adhesion of muscle skeletal cells and cooperates with FGF2 and IGF-1 to promote proliferation and survival. Cell Commun Adhes 12:41–57. https://doi.org/10.1080/15419060500383069
Lafont J, Jacques C, Le Dreau G, Calhabeu F, Thibout H, Dubois C, Berenbaum F, Laurent M, Martinerie C (2005b) New target genes for NOV/CCN3 in chondrocytes: TGF-beta2 and type X collagen. J Bone Miner Res 20:2213–2223. https://doi.org/10.1359/JBMR.050818
Lambi AG, Pankratz TL, Mundy C, Gannon M, Barbe MF, Richtsmeier JT, Popoff SN (2012) The skeletal site-specific role of connective tissue growth factor in prenatal osteogenesis. Dev Dyn 241:1944–1959. https://doi.org/10.1002/dvdy.23888
Lee CH, Shah B, Moioli EK, Mao JJ (2010a) CTGF directs fibroblast differentiation from human mesenchymal stem/stromal cells and defines connective tissue healing in a rodent injury model. J Clin Invest 120:3340–3349. https://doi.org/10.1172/JCI43230
Lee CH, Lee FY, Tarafder S, Kao K, Jun Y, Yang G, Mao JJ (2015) Harnessing endogenous stem/progenitor cells for tendon regeneration. J Clin Invest 125:2690–2701. https://doi.org/10.1172/JCI81589
Lee Y-T, Shao H-J, Wang J-H, Liu H-C, Hou S-M, Young T-H (2010b) Hyaluronic acid modulates gene expression of connective tissue growth factor (CTGF), transforming growth factor-β1 (TGF-β1), and vascular endothelial growth factor (VEGF) in human fibroblast-like synovial cells from advanced-stage osteoarthritis in vitro: hyaluronic acid modulates gene expression in human fibroblast-like synovial cellS. J Orthop Res 28:492–496. https://doi.org/10.1002/jor.21029
Li L, Kong X, Zang M, Hu B, Fang X, Gui B, Hu Y (2020) MicroRNA-584 impairs cellular proliferation and sensitizes osteosarcoma cells to cisplatin and taxanes by targeting CCN2. Cancer Manage Res 12:2577–2587. https://doi.org/10.2147/CMAR.S246545
Li X, Pongkitwitoon S, Lu H, Lee C, Gelberman R, Thomopoulos S (2019) CTGF induces tenogenic differentiation and proliferation of adipose-derived stromal cells. J Orthop Res 37:574–582. https://doi.org/10.1002/jor.24248
Li Z, Dang J, Chang KY, Rana TM (2014) MicroRNA-mediated regulation of extracellular matrix formation modulates somatic cell reprogramming. RNA 20:1900–1915. https://doi.org/10.1261/rna.043745.113
Lienau J, Schell H, Epari DR, Schütze N, Jakob F, Duda GN, Bail HJ (2006) CYR61 (CCN1) protein expression during fracture healing in an ovine tibial model and its relation to the mechanical fixation stability. J Orthop Res 24:254–262. https://doi.org/10.1002/jor.20035
Lin J, Zhou Z, Huo R, Xiao L, Ouyang G, Wang L, Sun Y, Shen B, Li D, Li N (2012) Cyr61 induces IL-6 production by fibroblast-like synoviocytes promoting Th17 differentiation in rheumatoid arthritis. J Immunol 188:5776–5784. https://doi.org/10.4049/jimmunol.1103201
Liu H, Peng F, Liu Z, Jiang F, Li L, Gao S, Wang G, Song J, Ruan E, Shao Z, Fu R (2017a) CYR61/CCN1 stimulates proliferation and differentiation of osteoblasts in vitro and contributes to bone remodelling in vivo in myeloma bone disease. Int J Oncol 50:631–639. https://doi.org/10.3892/ijo.2016.3815
Liu J, Tao X, Chen L, Han W, Zhou Y, Tang K (2015a) CTGF positively regulates BMP12 induced tenogenic differentiation of tendon stem cells and signaling. Cell Physiol Biochem 35:1831–1845. https://doi.org/10.1159/000373994
Liu S-C, Lee H-P, Hung C-Y, Tsai C-H, Li T-M, Tang C-H (2015b) Berberine attenuates CCN2-induced IL-1β expression and prevents cartilage degradation in a rat model of osteoarthritis. Toxicol Appl Pharmacol 289:20–29. https://doi.org/10.1016/j.taap.2015.08.020
Liu Y, Zhang F, Zhang Z, Wang D, Cui B, Zeng F, Huang L, Zhang Q, Sun Q (2017b) High expression levels of Cyr61 and VEGF are associated with poor prognosis in osteosarcoma. Pathol - Res Pract 213:895–899. https://doi.org/10.1016/j.prp.2017.06.004
MacDonald IJ, Huang C-C, Liu S-C, Lin Y-Y, Tang C-H (2021) Targeting CCN proteins in rheumatoid arthritis and osteoarthritis. Int J Mol Sci. https://doi.org/10.3390/ijms22094340
Madej W, van Caam A, Blaney Davidson EN, Hannink G, Buma P, van der Kraan PM (2016) Ageing is associated with reduction of mechanically-induced activation of Smad2/3P signaling in articular cartilage. Osteoarthr Cartil 24:146–157. https://doi.org/10.1016/j.joca.2015.07.018
Maeda A, Nishida T, Aoyama E, Kubota S, Lyons KM, Kuboki T, Takigawa M (2009) CCN family 2/connective tissue growth factor modulates BMP signalling as a signal conductor, which action regulates the proliferation and differentiation of chondrocytes. J Biochem 145:207–216. https://doi.org/10.1093/jb/mvn159
Maeda A, Ono M, Holmbeck K, Li L, Kilts TM, Kram V, Noonan ML, Yoshioka Y, McNerny EM, Tantillo MA, Kohn DH, Lyons KM, Robey PG, Young MF (2015) WNT1-induced secreted protein-1 (WISP1), a novel regulator of bone turnover and wnt signaling. J Biol Chem 290:14004–14018. https://doi.org/10.1074/jbc.M114.628818
Maeda-Uematsu A, Kubota S, Kawaki H, Kawata K, Miyake Y, Hattori T, Nishida T, Moritani N, Lyons KM, Iida S, Takigawa M (2014) CCN2 as a novel molecule supporting energy metabolism of chondrocytes. J Cell Biochem 115:854–865. https://doi.org/10.1002/jcb.24728
Magnusson C, Svensson A, Christerson U, Tågerud S (2005) Denervation-induced alterations in gene expression in mouse skeletal muscle. Eur J Neurosci 21:577–580. https://doi.org/10.1111/j.1460-9568.2005.03855.x
Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, Goldring SR, Jones G, Teichtahl AJ, Pelletier J-P (2016) Osteoarthritis. Nat Rev Disease Primers 2:16072. https://doi.org/10.1038/nrdp.2016.72
Matsushita Y, Sakamoto K, Tamamura Y, Shibata Y, Minamizato T, Kihara T, Ito M, Katsube K, Hiraoka S, Koseki H, Harada K, Yamaguchi A (2013) CCN3 protein participates in bone regeneration as an inhibitory factor. J Biol Chem 288:19973–19985. https://doi.org/10.1074/jbc.M113.454652
Matta A, Karim MZ, Gerami H, Jun P, Funabashi M, Kawchuk G, Goldstein A, Foltz W, Sussman M, Eek BC, Erwin WM (2018) NTG-101: a novel molecular therapy that halts the progression of degenerative disc disease. Sci Rep 8:16809. https://doi.org/10.1038/s41598-018-35011-4
Minamizato T, Sakamoto K, Liu T, Kokubo H, Katsube K, Perbal B, Nakamura S, Yamaguchi A (2007) CCN3/NOV inhibits BMP-2-induced osteoblast differentiation by interacting with BMP and Notch signaling pathways. Biochem Biophys Res Commun 354:567–573. https://doi.org/10.1016/j.bbrc.2007.01.029
Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF (2002) CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol 22:8709–8720. https://doi.org/10.1128/mcb.22.24.8709-8720.2002
Morales MG, Acuña MJ, Cabrera D, Goldschmeding R, Brandan E (2018) The pro-fibrotic connective tissue growth factor (CTGF/CCN2) correlates with the number of necrotic-regenerative foci in dystrophic muscle. J Cell Commun Signal 12:413–421. https://doi.org/10.1007/s12079-017-0409-3
Morales MG, Cabello-Verrugio C, Santander C, Cabrera D, Goldschmeding R, Brandan E (2011) CTGF/CCN-2 over-expression can directly induce features of skeletal muscle dystrophy. J Pathol 225:490–501. https://doi.org/10.1002/path.2952
Morales MG, Gutierrez J, Cabello-Verrugio C, Cabrera D, Lipson KE, Goldschmeding R, Brandan E (2013) Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum Mol Genet 22:4938–4951. https://doi.org/10.1093/hmg/ddt352
Moritani NH, Kubota S, Sugahara T, Takigawa M (2005) Comparable response of ccn1 with ccn2 genes upon arthritis: an in vitro evaluation with a human chondrocytic cell line stimulated bioset of cytokines. Cell Commun Signal 3:6. https://doi.org/10.1186/1478-811X-3-6
Murase Y, Hattori T, Aoyama E, Nishida T, Maeda-Uematsu A, Kawaki H, Lyons KM, Sasaki A, Takigawa M, Kubota S (2016) Role of CCN2 in amino acid metabolism of chondrocytes. J Cell Biochem 117:927–937. https://doi.org/10.1002/jcb.25377
Nakanishi T, Kimura Y, Tamura T, Ichikawa H, Yamaai Y, Sugimoto T, Takigawa M (1997) Cloning of a mRNA preferentially expressed in chondrocytes by differential display-PCR from a human chondrocytic cell line that is identical with connective tissue growth factor (CTGF) mRNA. Biochem Biophys Res Commun 234: 206–210. https://doi.org/10.1006/bbrc.1997.6528
Nakanishi T, Nishida T, Shimo T, Kobayashi K, Kubo T, Tamatani T, Tezuka K, Takigawa M (2000) Effects of CTGF/Hcs24, a product of a hypertrophic chondrocyte-specific gene, on the proliferation and differentiation of chondrocytes in culture. Endocrinology 141:264–273. https://doi.org/10.1210/endo.141.1.7267
Nakao K, Kubota S, Doi H, Eguchi T, Oka M, Fujisawa T, Nishida T, Takigawa M (2005) Collaborative action of M-CSF and CTGF/CCN2 in articular chondrocytes: possible regenerative roles in articular cartilage metabolism. Bone 36:884–892. https://doi.org/10.1016/j.bone.2004.10.015
Ni Y, Zhang H, Li Z (2020) Connective tissue growth factor (CCN2) inhibits TNF-α-induced apoptosis by enhancing autophagy through the Akt and Erk pathways in osteoblasts. Pharmazie 75:213–217. https://doi.org/10.1691/ph.2020.0336
Nishida T, Kubota S (2020) Roles of CCN2 as a mechano-sensing regulator of chondrocyte differentiation. Jpn Dent Sci Rev 56:119–126. https://doi.org/10.1016/j.jdsr.2020.07.001
Nishida T, Maeda A, Kubota S, Takigawa M (2008) Role of mechanical-stress inducible protein Hcs24/CTGF/CCN2 in cartilage growth and regeneration: mechanical stress induces expression of Hcs24/CTGF/CCN2 in a human chondrocytic cell line HCS-2/8, rabbit costal chondrocytes and meniscus tissue cells. Biorheology 45:289–299. https://doi.org/10.3233/BIR-2008-0478
Nishida T, Nakanishi T, Asano M, Shimo T, Takigawa M (2000) Effects of CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, on the proliferation and differentiation of osteoblastic cells in vitro. J Cell Physiol 184:197–206. https://doi.org/10.1002/1097-4652(200008)184:2%3c197::AID-JCP7%3e3.0.CO;2-R
Nishida T, Emura K, Kubota S, Lyons KM, Takigawa M (2011) CCN family 2/connective tissue growth factor (CCN2/CTGF) promotes osteoclastogenesis via induction of and interaction with dendritic cell-specific transmembrane protein (DC-STAMP). J Bone Miner Res 26:351–363. https://doi.org/10.1002/jbmr.222
Nishida T, Kubota S, Yokoi H, Mukoyama M, Takigawa M (2019) Roles of matricellular CCN2 deposited by osteocytes in osteoclastogenesis and osteoblast differentiation. Sci Rep 9:10913. https://doi.org/10.1038/s41598-019-47285-3
Nishida T, Kawaki H, Baxter RM, Deyoung RA, Takigawa M, Lyons KM (2007) CCN2 (Connective Tissue Growth Factor) is essential for extracellular matrix production and integrin signaling in chondrocytes. J Cell Commun Signal 1:45–58. https://doi.org/10.1007/s12079-007-0005-z
Nishida T, Kondo S, Maeda A, Kubota S, Lyons KM, Takigawa M (2009) CCN family 2/connective tissue growth factor (CCN2/CTGF) regulates the expression of Vegf through Hif-1alpha expression in a chondrocytic cell line, HCS-2/8, under hypoxic condition. Bone 44:24–31. https://doi.org/10.1016/j.bone.2008.08.125
Nishida T, Kubota S, Aoyama E, Janune D, Lyons KM, Takigawa M (2015) CCN family protein 2 (CCN2) promotes the early differentiation, but inhibits the terminal differentiation of skeletal myoblasts. J Biochem 157:91–100. https://doi.org/10.1093/jb/mvu056
Nishida T, Kubota S, Nakanishi T, Kuboki T, Yosimichi G, Kondo S, Takigawa M (2002) CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, stimulates proliferation and differentiation, but not hypertrophy of cultured articular chondrocytes. J Cell Physiol 192:55–63. https://doi.org/10.1002/jcp.10113
Nishida T, Kubota S, Kojima S, Kuboki T, Nakao K, Kushibiki T, Tabata Y, Takigawa M (2004) Regeneration of defects in articular cartilage in rat knee joints by CCN2 (Connective Tissue Growth Factor). J Bone Miner Res 19:1308–1319. https://doi.org/10.1359/JBMR.040322
Nozawa K, Fujishiro M, Kawasaki M, Kaneko H, Iwabuchi K, Yanagida M, Suzuki F, Miyazawa K, Takasaki Y, Ogawa H, Takamori K, Sekigawa I (2009) Connective tissue growth factor promotes articular damage by increased osteoclastogenesis in patients with rheumatoid arthritis. Arthritis Res Ther 11:R174. https://doi.org/10.1186/ar2863
Nozawa K (2014) Inhibition of rheumatoid arthritis by blocking connective tissue growth factor. World Journal of Orthopedics 5:653. https://doi.org/10.5312/wjo.v5.i5.653
Nozawa K, Fujishiro M, Kawasaki M, Yamaguchi A, Ikeda K, Morimoto S, Iwabuchi K, Yanagida M, Ichinose S, Morioka M, Ogawa H, Takamori K, Takasaki Y, Sekigawa I (2013) Inhibition of connective tissue growth factor ameliorates disease in a murine model of rheumatoid arthritis: blockade of CTGF ameliorates arthritis in mice with CIA. Arthritis Rheum 65:1477–1486. https://doi.org/10.1002/art.37902
O’Brien TP, Lau LF (1992) Expression of the growth factor-inducible immediate early gene cyr61 correlates with chondrogenesis during mouse embryonic development. Cell Growth Differ 3:645–654
O’Connor RS, Mills ST, Jones KA, Ho SN, Pavlath GK (2007) A combinatorial role for NFAT5 in both myoblast migration and differentiation during skeletal muscle myogenesis. J Cell Sci 120:149–159. https://doi.org/10.1242/jcs.03307
Ohgawara T, Kubota S, Kawaki H, Kondo S, Eguchi T, Kurio N, Aoyama E, Sasaki A, Takigawa M (2009) Regulation of chondrocytic phenotype by micro RNA 18a: involvement of Ccn2/Ctgf as a major target gene. FEBS Lett 583:1006–1010. https://doi.org/10.1016/j.febslet.2009.02.025
Ohgawara T, Kubota S, Kawaki H, Kurio N, Abd El Kader T, Hoshijima M, Janune D, Shimo T, Perbal B, Sasaki A, Takigawa M (2011) Association of the metastatic phenotype with CCN family members among breast and oral cancer cells. J Cell Commun Signal 5:291–299. https://doi.org/10.1007/s12079-011-0133-3
O’Kelly J, Chung A, Lemp N, Chumakova K, Yin D, Wang H-J, Said J, Gui D, Miller CW, Karlan BY, Koeffler HP (2008) Functional domains of CCN1 (Cyr61) regulate breast cancer progression. Int J Oncol 33:59–67
Ono M, Inkson CA, Sonn R, Kilts TM, de Castro LF, Maeda A, Fisher LW, Robey PG, Berendsen AD, Li L, McCartney-Francis N, Brown AC, Crawford NPS, Molinolo A, Jain A, Fedarko NS, Young MF (2013) WISP1/CCN4: a potential target for inhibiting prostate cancer growth and spread to bone. PLoS ONE 8:e71709. https://doi.org/10.1371/journal.pone.0071709
Pal A, Huang W, Li X, Toy KA, Nikolovska-Coleska Z, Kleer CG (2012) CCN6 modulates BMP signaling via the smad-independent TAK1/p38 pathway, acting to suppress metastasis of breast cancer. Can Res 72:4818–4828. https://doi.org/10.1158/0008-5472.CAN-12-0154
Parada C, Li J, Iwata J, Suzuki A, Chai Y (2013) CTGF mediates Smad-dependent transforming growth factor β signaling to regulate mesenchymal cell proliferation during palate development. Mol Cell Biol 33:3482–3493. https://doi.org/10.1128/MCB.00615-13
Perbal B (2004) CCN proteins: multifunctional signalling regulators. Lancet 363:62–64. https://doi.org/10.1016/S0140-6736(03)15172-0
Perbal B (2018) The concept of the CCN protein family revisited: a centralized coordination network. J Cell Commun Signal 12:3–12. https://doi.org/10.1007/s12079-018-0455-5
Perbal B, Lazar N, Zambelli D, Lopez-Guerrero JA, Llombart-Bosch A, Scotlandi K, Picci P (2009) Prognostic relevance of CCN3 in Ewing sarcoma. Hum Pathol 40:1479–1486. https://doi.org/10.1016/j.humpath.2009.05.008
Perbal B, Zuntini M, Zambelli D, Serra M, Sciandra M, Cantiani L, Lucarelli E, Picci P, Scotlandi K (2008) Prognostic value of CCN3 in osteosarcoma. Clin Cancer Res 14:701–709. https://doi.org/10.1158/1078-0432.CCR-07-0806
Pereira RC, Durant D, Canalis E (2000) Transcriptional regulation of connective tissue growth factor by cortisol in osteoblasts. Am J Physiol Endocrinol Metab 279:E570–E576. https://doi.org/10.1152/ajpendo.2000.279.3.E570
Petrosino JM, Leask A, Accornero F (2019) Genetic manipulation of CCN2/CTGF unveils cell-specific ECM-remodeling effects in injured skeletal muscle. FASEB J 33:2047–2057. https://doi.org/10.1096/fj.201800622RR
Rachfal AW, Brigstock DR (2005) Structural and functional properties of CCN proteins. Vitam Harmon 70:69–103. https://doi.org/10.1016/S0083-6729(05)70003-0
Rao H, Wu Z, Wei S, Jiang Y, Guo Q, Wang J, Chen C, Yang H (2020) MiR-25-3p serves as an oncogenic MicroRNA by downregulating the expression of merlin in osteosarcoma. Cancer Manag Res 12:8989–9001. https://doi.org/10.2147/CMAR.S262245
Rebolledo DL, Acuña MJ, Brandan E (2021) Role of matricellular CCN proteins in skeletal muscle: focus on CCN2/CTGF and its regulation by vasoactive peptides. Int J Mol Sci. https://doi.org/10.3390/ijms22105234
Reed DR, Hayashi M, Wagner L, Binitie O, Steppan DA, Brohl AS, Shinohara ET, Bridge JA, Loeb DM, Borinstein SC, Isakoff MS (2017) Treatment pathway of bone sarcoma in children, adolescents, and young adults: consensus bone sarcoma treatment pathway. Cancer 123:2206–2218. https://doi.org/10.1002/cncr.30589
Ren W, Sun X, Wang K, Feng H, Liu Y, Fei C, Wan S, Wang W, Luo J, Shi Q, Tang M, Zuo G, Weng Y, He T, Zhang Y (2014) BMP9 inhibits the bone metastasis of breast cancer cells by downregulating CCN2 (connective tissue growth factor, CTGF) expression. Mol Biol Rep 41:1373–1383. https://doi.org/10.1007/s11033-013-2982-8
Repudi SR, Patra M, Sen M (2013) WISP3-IGF1 interaction regulates chondrocyte hypertrophy. J Cell Sci 126:1650–1658. https://doi.org/10.1242/jcs.119859
Roddy KA, Boulter CA (2015) Targeted mutation of NOV/CCN3 in mice disrupts joint homeostasis and causes osteoarthritis-like disease. Osteoarthr Cartil 23:607–615. https://doi.org/10.1016/j.joca.2014.12.012
Rui Y, Chen M, Li Y, Xiao L, Geng P, Wang P, Xu Z, Zhang X, Dai G (2019) CTGF Attenuates Tendon-Derived Stem/Progenitor Cell Aging. Stem Cells Int 2019:1–12. https://doi.org/10.1155/2019/6257537
Rydziel S, Stadmeyer L, Zanotti S, Durant D, Smerdel-Ramoya A, Canalis E (2007) Nephroblastoma overexpressed (Nov) inhibits osteoblastogenesis and causes osteopenia. J Biol Chem 282:19762–19772. https://doi.org/10.1074/jbc.M700212200
Safadi FF, Xu J, Smock SL, Kanaan RA, Selim AH, Odgren PR, Marks SC, Owen TA, Popoff SN (2003) Expression of connective tissue growth factor in bone: its role in osteoblast proliferation and differentiation in vitro and bone formation in vivo. J Cell Physiol 196:51–62. https://doi.org/10.1002/jcp.10319
Sakamoto K, Yamaguchi S, Ando R, Miyawaki A, Kabasawa Y, Takagi M, Li CL, Perbal B, Katsube K (2002) The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with notch1 extracellular domain and inhibits myoblast differentiation via notch signaling pathway. J Biol Chem 277:29399–29405. https://doi.org/10.1074/jbc.M203727200
Salhotra A, Shah HN, Levi B, Longaker MT (2020) Mechanisms of bone development and repair. Nat Rev Mol Cell Biol 21:696–711. https://doi.org/10.1038/s41580-020-00279-w
Samarpita S, Ganesan R, Rasool M (2020) Cyanidin prevents the hyperproliferative potential of fibroblast-like synoviocytes and disease progression via targeting IL-17A cytokine signalling in rheumatoid arthritis. Toxicol Appl Pharmacol 391:114917. https://doi.org/10.1016/j.taap.2020.114917
Schlegelmilch K, Keller A, Zehe V, Hondke S, Schilling T, Jakob F, Klein-Hitpass L, Schütze N (2014) WISP 1 is an important survival factor in human mesenchymal stromal cells. Gene 551:243–254. https://doi.org/10.1016/j.gene.2014.09.002
Schutze N, Noth U, Schneidereit J, Hendrich C, Jakob F (2005) Differential expression of CCN-family members in primary human bone marrow-derived mesenchymal stem cells during osteogenic, chondrogenic and adipogenic differentiation. Cell Commun Signal 3:5. https://doi.org/10.1186/1478-811X-3-5
Sen M, Cheng YH, Goldring MB, Lotz MK, Carson DA (2004) WISP3-dependent regulation of type II collagen and aggrecan production in chondrocytes. Arthritis Rheum 50:488–497. https://doi.org/10.1002/art.20005
Shakunaga T, Ozaki T, Ohara N, Asaumi K, Doi T, Nishida K, Kawai A, Nakanishi T, Takigawa M, Inoue H (2000) Expression of connective tissue growth factor in cartilaginous tumors. Cancer 89:1466–1473
Shimo T, Kubota S, Yoshioka N, Ibaragi S, Isowa S, Eguchi T, Sasaki A, Takigawa M (2006) Pathogenic role of connective tissue growth factor (CTGF/CCN2) in osteolytic metastasis of breast cancer. J Bone Miner Res 21:1045–1059. https://doi.org/10.1359/jbmr.060416
Shimo T, Kubota S, Goda T, Yoshihama Y, Kurio N, Nishida T, Ng P-S, Endo K, Takigawa M, Sasakii A (2008) Clinical significance and pathogenic function of connective tissue growth factor (CTGF/CCN2) in osteolytic mandibular squamous cell carcinoma. Anticancer Res 28:2343–2348
Siegel RL, Miller KD, Fuchs HE, Jemal A (2021) Cancer Statistics. CA A Cancer J Clin 71:7–33. https://doi.org/10.3322/caac.21654
Smerdel-Ramoya A, Zanotti S, Deregowski V, Canalis E (2008) Connective tissue growth factor enhances osteoblastogenesis in vitro. J Biol Chem 283:22690–22699. https://doi.org/10.1074/jbc.M710140200
Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, Kavanaugh A, McInnes IB, Solomon DH, Strand V, Yamamoto K (2018) Rheumatoid arthritis. Nat Rev Disease Primers 4:18001. https://doi.org/10.1038/nrdp.2018.1
Song JJ, Aswad R, Kanaan RA, Rico MC, Owen TA, Barbe MF, Safadi FF, Popoff SN (2007) Connective tissue growth factor (CTGF) acts as a downstream mediator of TGF-beta1 to induce mesenchymal cell condensation. J Cell Physiol 210:398–410. https://doi.org/10.1002/jcp.20850
Song Y, Yao S, Liu Y, Long L, Yang H, Li Q, Liang J, Li X, Lu Y, Zhu H, Zhang N (2017) Expression levels of TGF-β1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp Ther Med 13:1209–1214. https://doi.org/10.3892/etm.2017.4105
Strammiello R, Benini S, Manara MC, Perdichizzi S, Serra M, Spisni E, Picci P, Scotlandi K (2003) Impact of IGF-I/IGF-IR circuit on the angiogenetic properties of Ewing’s sarcoma cells. Hormone and metabolic research hormon Und Stoffwechselforschung. Hormon Et Metab 35:675–684. https://doi.org/10.1055/s-2004-814149
Sumiyoshi K, Kubota S, Ohgawara T, Kawata K, Abd El Kader T, Nishida T, Ikeda N, Shimo T, Yamashiro T, Takigawa M (2013) Novel role of miR-181a in cartilage metabolism. J Cell Biochem 114:2094–2100. https://doi.org/10.1002/jcb.24556
Sun G, Haginoya K, Wu Y, Chiba Y, Nakanishi T, Onuma A, Sato Y, Takigawa M, Iinuma K, Tsuchiya S (2008) Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J Neurol Sci 267:48–56. https://doi.org/10.1016/j.jns.2007.09.043
Sun W, Ma J, Zhao H, Xiao C, Zhong H, Ling H, Xie Z, Tian Q, Chen H, Zhang T, Chen M, Jin S, Wang J (2020) Resolvin D1 suppresses pannus formation via decreasing connective tissue growth factor caused by upregulation of miRNA-146a-5p in rheumatoid arthritis. Arthritis Res Ther 22:61. https://doi.org/10.1186/s13075-020-2133-2
Tai HC, Chang AC, Yu HJ, Huang CY, Tsai YC, Lai YW, Sun HL, Tang CH, Wang SW (2014) Osteoblast-derived increases expression and enhances prostate cancer metastasis by down-regulating. Oncotarget 5:7589–7598. https://doi.org/10.18632/oncotarget.2280
Tan T-W, Lai C-H, Huang C-Y, Yang W-H, Chen H-T, Hsu H-C, Fong Y-C, Tang C-H (2009a) CTGF enhances migration and MMP-13 up-regulation via αvβ3 integrin, FAK, ERK, and NF-κB-dependent pathway in human chondrosarcoma cells. J Cell Biochem 107:345–356. https://doi.org/10.1002/jcb.22132
Tan T-W, Yang W-H, Lin Y-T, Hsu S-F, Li T-M, Kao S-T, Chen W-C, Fong Y-C, Tang C-H (2009b) Cyr61 increases migration and MMP-13 expression via αvβ3 integrin, FAK, ERK and AP-1-dependent pathway in human chondrosarcoma cells. Carcinogenesis 30:258–268. https://doi.org/10.1093/carcin/bgn284
Tarafder S, Chen E, Jun Y, Kao K, Sim KH, Back J, Lee FY, Lee CH (2017) Tendon stem/progenitor cells regulate inflammation in tendon healing. FASEB J 31:3991–3998. https://doi.org/10.1096/fj.201700071R
Torreggiani S, Torcoletti M, Campos-Xavier B, Baldo F, Agostoni C, Superti-Furga A, Filocamo G (2019) Progressive pseudorheumatoid dysplasia: a rare childhood disease. Rheumatol Int 39:441–452. https://doi.org/10.1007/s00296-018-4170-6
Tran MN, Kleer CG (2018) Matricellular CCN6 (WISP3) protein: a tumor suppressor for mammary metaplastic carcinomas. J Cell Commun Signal 12:13–19. https://doi.org/10.1007/s12079-018-0451-9
Tsai HC, Huang CY, Su HL, Tang CH (2014) CCN2 enhances resistance to cisplatin-mediating cell apoptosis in human osteosarcoma. PLoS ONE 9:e90159. https://doi.org/10.1371/journal.pone.0090159
Tsai HC, Huang CY, Su HL, Tang CH (2014) CTGF increases drug resistance to paclitaxel by upregulating survivin expression in human osteosarcoma cells. Biochimica et Biophysica Acta BBA Mole Cell Res 1843:846–854. https://doi.org/10.1016/j.bbamcr.2014.01.007
Tsai HC, Su HL, Huang CY, Fong YC, Hsu CJ, Tang CH (2014) CTGF increases matrix metalloproteinases expression and subsequently promotes tumor metastasis in human osteosarcoma through down-regulating miR-519d. Oncotarget 5:3800–3812. https://doi.org/10.18632/oncotarget.1998
Tsai H-C, Tzeng H-E, Huang C-Y, Huang Y-L, Tsai C-H, Wang S-W, Wang P-C, Chang A-C, Fong Y-C, Tang C-H (2017) WISP-1 positively regulates angiogenesis by controlling VEGF-A expression in human osteosarcoma. Cell Death Dis 8:e2750–e2750. https://doi.org/10.1038/cddis.2016.421
Tzeng H-E, Chen J-C, Tsai C-H, Kuo C-C, Hsu H-C, Hwang W-L, Fong Y-C, Tang C-H (2011) CCN3 increases cell motility and MMP-13 expression in human chondrosarcoma through integrin-dependent pathway. J Cell Physiol 226:3181–3189. https://doi.org/10.1002/jcp.22672
Urano T, Narusawa K, Shiraki M, Usui T, Sasaki N, Hosoi T, Ouchi Y, Nakamura T, Inoue S (2007) Association of a single nucleotide polymorphism in the WISP1 gene with spinal osteoarthritis in postmenopausal Japanese women. J Bone Miner Metab 25:253–258. https://doi.org/10.1007/s00774-007-0757-9
van den Bosch MH, Blom AB, van Lent PL, van Beuningen HM, Blaney Davidson EN, van der Kraan PM, van den Berg WB (2014) Canonical Wnt signaling skews TGF-β signaling in chondrocytes towards signaling via ALK1 and Smad 1/5/8. Cell Signal 26:951–958. https://doi.org/10.1016/j.cellsig.2014.01.021
van den Bosch MH, Blom AB, Sloetjes AW, Koenders MI, van de Loo FA, van den Berg WB, van Lent PL, van der Kraan PM (2015) Induction of canonical wnt signaling by synovial overexpression of selected wnts leads to protease activity and early osteoarthritis-like cartilage damage. Am J Pathol 185:1970–1980. https://doi.org/10.1016/j.ajpath.2015.03.013
van den Bosch MH, Blom AB, Kram V, Maeda A, Sikka S, Gabet Y, Kilts TM, van den Berg WB, van Lent PL, van der Kraan PM, Young MF (2017) WISP1/CCN4 aggravates cartilage degeneration in experimental osteoarthritis. Osteoarthr Cartil 25:1900–1911. https://doi.org/10.1016/j.joca.2017.07.012
van den Bosch MHJ, Ramos YFM, den Hollander W, Bomer N, Nelissen RGHH, Bovée JVMG, van den Berg WB, van Lent PLEM, Blom AB, van der Kraan PM, Meulenbelt I (2019) Increased WISP1 expression in human osteoarthritic articular cartilage is epigenetically regulated and decreases cartilage matrix production. Rheumatology 58:1065–1074. https://doi.org/10.1093/rheumatology/key426
Wang H, Xu Q, Xiao F, Jiang Y, Wu Z (2008) Involvement of the p38 mitogen-activated protein kinase alpha, beta, and gamma isoforms in myogenic differentiation. Mol Biol Cell 19:1519–1528. https://doi.org/10.1091/mbc.e07-08-0817
Wang JJ, Ye F, Cheng LJ, Shi YJ, Bao J, Sun HQ, Wang W, Zhang P, Bu H (2009) Osteogenic differentiation of mesenchymal stem cells promoted by overexpression of connective tissue growth factor. J Zhejiang Univ Sci B 10:355–367. https://doi.org/10.1631/jzus.B0820252
Wang Y, Jiang L, Dai G, Li S, Mu X (2018) Bioinformatics analysis reveals different gene expression patterns in the annulus fibrosis and nucleus pulpous during intervertebral disc degeneration. Exp Ther Med. https://doi.org/10.3892/etm.2018.6884
Wei Y, Peng L, Li Y, Zhang N, Shang K, Duan L, Zhong J, Chen J (2020) Higher serum CCN3 Is associated with disease activity and inflammatory markers in rheumatoid arthritis. J Immunol Res 2020:1–7. https://doi.org/10.1155/2020/3891425
Witte F, Dokas J, Neuendorf F, Mundlos S, Stricker S (2009) Comprehensive expression analysis of all Wnt genes and their major secreted antagonists during mouse limb development and cartilage differentiation. Gene Expr Patterns 9:215–223. https://doi.org/10.1016/j.gep.2008.12.009
Wong M, Kireeva ML, Kolesnikova TV, Lau LF (1997) Cyr61, product of a growth factor-inducible immediate-early gene, regulates chondrogenesis in mouse limb bud mesenchymal cells. Dev Biol 192:492–508. https://doi.org/10.1006/dbio.1997.8766
Woods A, Pala D, Kennedy L, McLean S, Rockel JS, Wang G, Leask A, Beier F (2009) Rac1 signaling regulates CTGF/CCN2 gene expression via TGFbeta/Smad signaling in chondrocytes. Osteoarthr Cartil 17:406–413. https://doi.org/10.1016/j.joca.2008.07.002
Wu C-L, Tsai H-C, Chen Z-W, Wu C-M, Li T-M, Fong Y-C, Tang C-H (2013) Ras activation mediates WISP-1-induced increases in cell motility and matrix metalloproteinase expression in human osteosarcoma. Cell Signal 25:2812–2822. https://doi.org/10.1016/j.cellsig.2013.09.005
Xiang Z, Zeng Z, Tang Z, Fan J, He J, Zeng H, Zhu X (2011) Potential prognostic biomarkers for bone metastasis from hepatocellular carcinoma. Oncologist 16:1028–1039. https://doi.org/10.1634/theoncologist.2010-0358
Xiang Z-L, Zeng Z-C, Fan J, Tang Z-Y, Zeng H-Y (2012) Expression of connective tissue growth factor and interleukin-11 in intratumoral tissue is associated with poor survival after curative resection of hepatocellular carcinoma. Mol Biol Rep 39:6001–6006. https://doi.org/10.1007/s11033-011-1413-y
Xing X, Li Z, Yu Z, Cheng G, Li D (2018) Effects of connective tissue growth factor (CTGF/CCN2) on condylar chondrocyte proliferation, migration, maturation, differentiation and signalling pathway. Biochem Biophys Res Commun 495:1447–1453. https://doi.org/10.1016/j.bbrc.2017.11.190
Xu F, Teitelbaum SL (2013) Osteoclasts: new insights. Bone Res 1:11–26. https://doi.org/10.4248/BR201301003
Xu Y, Chu H, Zhou Y, Wang J, Dong C, Yin R (2018) miR-365 functions as a tumor suppressor by directly targeting CYR61 in osteosarcoma. Biomed Pharmacother 98:531–537. https://doi.org/10.1016/j.biopha.2017.12.086
Yamamoto Y, Furukawa K, Ueyama K, Nakanishi T, Takigawa M, Harata S (2002) Possible roles of CTGF/Hcs24 in the initiation and development of ossification of the posterior longitudinal ligament. Spine Phila Pa 1976 27:1852–7. https://doi.org/10.1097/00007632-200209010-00009
Yanagita T, Kubota S, Kawaki H, Kawata K, Kondo S, Takano-Yamamoto T, Tanaka S, Takigawa M (2007) Expression and physiological role of CCN4/Wnt-induced secreted protein 1 mRNA splicing variants in chondrocytes. FEBS J 274:1655–1665. https://doi.org/10.1111/j.1742-4658.2007.05709.x
Yang X, Lin K, Ni S, Wang J, Tian Q, Chen H, Brown MA, Zheng K, Zhai W, Sun L, Jin S, Wang J (2017) Serum connective tissue growth factor is a highly discriminatory biomarker for the diagnosis of rheumatoid arthritis. Arthritis Res Ther 19:257. https://doi.org/10.1186/s13075-017-1463-1
Yao J, Weng Y, Yan S, Hou M, Wang H, Shi Q, Zuo G (2015) NOV inhibits proliferation while promoting apoptosis and migration in osteosarcoma cell lines through p38/MAPK and JNK/MAPK pathways. Oncol Rep 34:2011–2021. https://doi.org/10.3892/or.2015.4153
Yeger H, Perbal B (2021) The CCN axis in cancer development and progression. J Cell Commun Signal. https://doi.org/10.1007/s12079-021-00618-2
Yoshioka Y, Ono M, Maeda A, Kilts TM, Hara ES, Khattab H, Ueda J, Aoyama E, Oohashi T, Takigawa M, Young MF, Kuboki T (2016) CCN4/WISP-1 positively regulates chondrogenesis by controlling TGF-β3 function. Bone 83:162–170. https://doi.org/10.1016/j.bone.2015.11.007
Yosimichi G, Nakanishi T, Nishida T, Hattori T, Takano-Yamamoto T, Takigawa M (2001) CTGF/Hcs24 induces chondrocyte differentiation through a p38 mitogen-activated protein kinase (p38MAPK), and proliferation through a p44/42 MAPK/extracellular-signal regulated kinase (ERK). Eur J Biochem 268:6058–6065. https://doi.org/10.1046/j.0014-2956.2001.02553.x
Yu C, Le AT, Yeger H, Perbal B, Alman BA (2003) NOV (CCN3) regulation in the growth plate and CCN family member expression in cartilage neoplasia. J Pathol 201:609–615. https://doi.org/10.1002/path.1468
Yu WW, Xia Q, Wu Y, Bu QY (2014) Activation of PPAR-γ inhibits differentiation of rat osteoblasts by reducing expression of connective tissue growth factor. J Huazhong Univ Sci Technolog Med Sci 34:652–656. https://doi.org/10.1007/s11596-014-1332-y
Yuan LQ, Lu Y, Luo XH, Xie H, Wu XP, Liao EY (2007) Taurine promotes connective tissue growth factor (CTGF) expression in osteoblasts through the ERK signal pathway. Amino Acids 32:425–430. https://doi.org/10.1007/s00726-006-0380-4
Zhai T, Gao C, Huo R, Sheng H, Sun S, Xie J, He Y, Gao H, Li H, Zhang J, Li H, Sun Y, Lin J, Shen B, Xiao L, Li N (2017) Cyr61 participates in the pathogenesis of rheumatoid arthritis via promoting MMP-3 expression by fibroblast-like synoviocytes. Mod Rheumatol 27:466–475. https://doi.org/10.1080/14397595.2016.1220447
Zhang H, Qu S, Li S, Wang Y, Li Y, Wang Y, Wang Z, Li R (2013) Silencing SATB1 inhibits proliferation of human osteosarcoma U2OS cells. Mol Cell Biochem 378:39–45. https://doi.org/10.1007/s11010-013-1591-0
Zhang L, Niu H, Ma J, Yuan B-Y, Chen Y-H, Zhuang Y, Chen G-W, Zeng Z-C, Xiang Z-L (2019) The molecular mechanism of LncRNA34a-mediated regulation of bone metastasis in hepatocellular carcinoma. Mol Cancer 18:120. https://doi.org/10.1186/s12943-019-1044-9
Zhang Q, Wu J, Cao Q, Xiao L, Wang L, He D, Ouyang G, Lin J, Shen B, Shi Y, Zhang Y, Li D, Li N (2009) A critical role of Cyr61 in interleukin-17–dependent roliferation of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Rheum 60:3602–3612. https://doi.org/10.1002/art.24999
Zhang S, Li B, Tang W, Ni L, Ma H, Lu M, Meng Q (2018) Effects of connective tissue growth factor on prostate cancer bone metastasis and osteoblast differentiation. Oncol Lett. https://doi.org/10.3892/ol.2018.8960
Zhao G, Huang BL, Rigueur D, Wang W, Bhoot C, Charles KR, Baek J, Mohan S, Jiang J, Lyons KM (2018) CYR61/CCN1 Regulates sclerostin levels and bone maintenance. J Bone Miner Res 33:1076–1089. https://doi.org/10.1002/jbmr.3394
Zhou M, Ze K, Hua L, Liu L, Kuai L, Zhang M, Li B, Wang Y, Li X (2020) Cyr61 Promotes Inflammation of a Gouty Arthritis Model in Rats. Mediators Inflamm 2020:1–13. https://doi.org/10.1155/2020/8298615
Zhu X, Song Y, Huo R, Zhang J, Sun S, He Y, Gao H, Zhang M, Sun X, Zhai T, Li H, Sun Y, Zhou Z, Shen B, Xiao L, Li N (2015) Cyr61 participates in the pathogenesis of rheumatoid arthritis by promoting proIL-1β production by fibroblast-like synoviocytes through an AKT-dependent NF-κB signaling pathway. Clin Immunol 157:187–197. https://doi.org/10.1016/j.clim.2015.02.010
Zieba J, Forlenza KN, Khatra JS, Sarukhanov A, Duran I, Rigueur D, Lyons KM, Cohn DH, Merrill AE, Krakow D (2016) TGFβ and BMP dependent cell fate changes due to loss of filamin B produces disc degeneration and progressive vertebral fusions. PLoS Genet 12:e1005936. https://doi.org/10.1371/journal.pgen.1005936
Acknowledgements
The authors thank Cristina Ghinelli for editing the figures. The authors would like to apologize to investigators whose work was not cited in this review due to space limitations.
Funding
The research leading to these results has received funding from AIRC under IG 2019 – ID. 22805 project – P.I. Scotlandi Katia.
Author information
Authors and Affiliations
Contributions
All authors have made a substantial, direct and intellectual contribution to the work and agreed to the published version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Giusti, V., Scotlandi, K. CCN proteins in the musculoskeletal system: current understanding and challenges in physiology and pathology. J. Cell Commun. Signal. 15, 545–566 (2021). https://doi.org/10.1007/s12079-021-00631-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-021-00631-5